lahuuki@gmail.com -
7 August 2024
+12 August 2024
Source:vignettes/DeconvoBuddies.Rmd
DeconvoBuddies.RmdAccess Data
## Access and snRNA-seq example data
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:45.703235 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:08:35.815188 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## Explore snRNA-seq data in sce_DLPFC_example
sce_DLPFC_example
@@ -239,7 +239,7 @@ Access Data## Access Bulk RNA-seq data
if (!exists("rse_gene")) rse_gene <- fetch_deconvo_data("rse_gene")
-#> 2024-08-07 17:33:47.478342 loading file /github/home/.cache/R/BiocFileCache/4a1de5efa9_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
+#> 2024-08-12 15:08:37.663146 loading file /github/home/.cache/R/BiocFileCache/146b2dbc692f_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
## Explore bulk data in rse_gene
rse_gene
@@ -470,9 +470,9 @@ Reproducibilitylibrary("knitr")
knit("DeconvoBuddies.Rmd", tangle = TRUE)
Date the vignette was generated.
-#> [1] "2024-08-07 17:33:51 UTC"
+#> [1] "2024-08-12 15:08:43 UTC"
Wallclock time spent generating the vignette.
-#> Time difference of 18.582 secs
+#> Time difference of 19.447 secs
R session information.
#> ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
#> setting value
@@ -484,8 +484,8 @@ Reproducibility#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz UTC
-#> date 2024-08-07
-#> pandoc 3.2 @ /usr/bin/ (via rmarkdown)
+#> date 2024-08-12
+#> pandoc 3.3 @ /usr/bin/ (via rmarkdown)
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
@@ -529,7 +529,7 @@ Reproducibility#> data.table 1.15.4 2024-03-30 [1] RSPM (R 4.4.0)
#> DBI 1.2.3 2024-06-02 [1] RSPM (R 4.4.0)
#> dbplyr 2.5.0 2024-03-19 [1] RSPM (R 4.4.0)
-#> DeconvoBuddies * 0.99.0 2024-08-07 [1] Bioconductor
+#> DeconvoBuddies * 0.99.0 2024-08-12 [1] Bioconductor
#> DelayedArray 0.31.11 2024-08-04 [1] Bioconductor 3.20 (R 4.4.1)
#> desc 1.4.3 2023-12-10 [2] RSPM (R 4.4.0)
#> digest 0.6.36 2024-06-23 [2] RSPM (R 4.4.0)
@@ -594,7 +594,7 @@ Reproducibility#> paletteer 1.6.0 2024-01-21 [1] RSPM (R 4.4.0)
#> pillar 1.9.0 2023-03-22 [2] RSPM (R 4.4.0)
#> pkgconfig 2.0.3 2019-09-22 [2] RSPM (R 4.4.0)
-#> pkgdown 2.1.0.9000 2024-08-05 [1] Github (r-lib/pkgdown@1d40a80)
+#> pkgdown 2.1.0.9000 2024-08-12 [1] Github (r-lib/pkgdown@1d40a80)
#> plotly 4.10.4 2024-01-13 [1] RSPM (R 4.4.0)
#> plyr 1.8.9 2023-10-02 [1] RSPM (R 4.4.0)
#> png 0.1-8 2022-11-29 [1] RSPM (R 4.4.0)
diff --git a/articles/Deconvolution_Benchmark_DLPFC.html b/articles/Deconvolution_Benchmark_DLPFC.html
index b842b41..d83f1e8 100644
--- a/articles/Deconvolution_Benchmark_DLPFC.html
+++ b/articles/Deconvolution_Benchmark_DLPFC.html
@@ -98,7 +98,7 @@ Louise
Campus
lahuuki@gmail.com
- 7 August 2024
+ 12 August 2024
Source: vignettes/Deconvolution_Benchmark_DLPFC.Rmd
Deconvolution_Benchmark_DLPFC.Rmd
@@ -138,46 +138,46 @@ Deconvolution Methods
+
Approach
Method
Citation
Availability
-
+
weighted least squares
DWLS
Tsoucas et al, Nature Comm, 2019
R
Package Cran
-
+
Bias correction: Assay
Bisque
Jew et al, Nature Comm, 2020
R Package github
-
+
Bias correction: Sourse
MuSiC
Wang et al, Nature Communications, 2019
R Package github
-
+
Machine Learning
CIBERSORTx
Newman et al., Nature BioTech, 2019
Webtool
-
+
Bayesian
BayesPrism
Chu et al., Nature Cancer, 2022
Webtool/R
Package
-
+
linear
Hspe
Hunt et al., Ann. Appl. Stat, 2021
@@ -261,7 +261,7 @@ Bulk RNA-seq data
## use fetch deconvon data to load rse_gene
rse_gene <- fetch_deconvo_data("rse_gene")
-#> 2024-08-07 17:34:07.847847 loading file /github/home/.cache/R/BiocFileCache/4a1de5efa9_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
+#> 2024-08-12 15:08:59.333378 loading file /github/home/.cache/R/BiocFileCache/146b2dbc692f_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
rse_gene
#> class: RangedSummarizedExperiment
#> dim: 21745 110
@@ -297,7 +297,7 @@ Reference snRNA-seq data
## Use spatialLIBD to fetch the snRNA-seq dataset
sce_path_zip <- fetch_deconvo_data("sce")
-#> 2024-08-07 17:34:09.528637 loading file /github/home/.cache/R/BiocFileCache/4a1415db437_sce_DLPFC_annotated.zip%3Fdl%3D1
+#> 2024-08-12 15:09:01.898358 loading file /github/home/.cache/R/BiocFileCache/146b2a8cb8a7_sce_DLPFC_annotated.zip%3Fdl%3D1
## unzip and load the data
sce_path <- unzip(sce_path_zip, exdir = tempdir())
@@ -388,10 +388,12 @@ 3. Select Marker Genes
We have developed a method for finding marker genes called the “Mean
-Ratio”. We calculate the Mean Ratio for a target cell type
+Ratio”. We calculate the MeanRatio for a target cell type
for each gene by dividing the mean expression of the target cell by the
mean expression of the next highest non-target cell type. Genes with the
-highest Mean Ratio values are selected as marker genes.
+highest MeanRatio values are selected as marker genes.
+For a tutorial on marker gene selection check out Vignette:
+Deconvolution Benchmark in Human DLPFC.
@@ -762,9 +764,9 @@ Reproducibilitylibrary("knitr")
knit("Deconvolution_Benchmark_DLPFC.Rmd", tangle = TRUE)
Date the vignette was generated.
-#> [1] "2024-08-07 17:35:08 UTC"
+#> [1] "2024-08-12 15:10:03 UTC"
Wallclock time spent generating the vignette.
-#> Time difference of 1.214 mins
+#> Time difference of 1.27 mins
R session information.
#> ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
#> setting value
@@ -776,8 +778,8 @@ Reproducibility#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz UTC
-#> date 2024-08-07
-#> pandoc 3.2 @ /usr/bin/ (via rmarkdown)
+#> date 2024-08-12
+#> pandoc 3.3 @ /usr/bin/ (via rmarkdown)
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
@@ -822,7 +824,7 @@ Reproducibility#> data.table 1.15.4 2024-03-30 [1] RSPM (R 4.4.0)
#> DBI 1.2.3 2024-06-02 [1] RSPM (R 4.4.0)
#> dbplyr 2.5.0 2024-03-19 [1] RSPM (R 4.4.0)
-#> DeconvoBuddies * 0.99.0 2024-08-07 [1] Bioconductor
+#> DeconvoBuddies * 0.99.0 2024-08-12 [1] Bioconductor
#> DelayedArray 0.31.11 2024-08-04 [1] Bioconductor 3.20 (R 4.4.1)
#> desc 1.4.3 2023-12-10 [2] RSPM (R 4.4.0)
#> digest 0.6.36 2024-06-23 [2] RSPM (R 4.4.0)
@@ -891,7 +893,7 @@ Reproducibility#> paletteer 1.6.0 2024-01-21 [1] RSPM (R 4.4.0)
#> pillar 1.9.0 2023-03-22 [2] RSPM (R 4.4.0)
#> pkgconfig 2.0.3 2019-09-22 [2] RSPM (R 4.4.0)
-#> pkgdown 2.1.0.9000 2024-08-05 [1] Github (r-lib/pkgdown@1d40a80)
+#> pkgdown 2.1.0.9000 2024-08-12 [1] Github (r-lib/pkgdown@1d40a80)
#> plotly 4.10.4 2024-01-13 [1] RSPM (R 4.4.0)
#> plyr 1.8.9 2023-10-02 [1] RSPM (R 4.4.0)
#> png 0.1-8 2022-11-29 [1] RSPM (R 4.4.0)
diff --git a/articles/Marker_Finding.html b/articles/Marker_Finding.html
index 54c0154..8626235 100644
--- a/articles/Marker_Finding.html
+++ b/articles/Marker_Finding.html
@@ -98,7 +98,7 @@ Louise
Campus
lahuuki@gmail.com
- 7 August 2024
+ 12 August 2024
Source: vignettes/Marker_Finding.Rmd
Marker_Finding.Rmd
@@ -113,21 +113,74 @@ Introduction
What are Marker Genes?
+Cell type marker genes have cell type specific expression, that is
+high expression in the target cell type, and low expression in all other
+cell types. Sub-setting the genes considered in a cell type
+deconvolution analysis helps reduce noise and can improve the accuracy
+of a deconvolution method.
How can we select marker genes?
+There are several approaches to select marker genes.
+One popular method is “1 vs. All” differential expression (Lun et
+al., 2016, F1000Res) (TODO update citation), where genes are tested for
+differential expression between the target cell type, and a combined
+group of all “other” cell types. Statistically
+significant differntially expressed genes (DEGs) can be selected as a
+set of marker genes, DEGs can be ranked by high log fold change.
+However in some cases 1vAll can select genes with high
+expression in non-target cell types, especially in cell types related to
+the target cell types (such as Neuron sub-types), or when there is a
+smaller number of cells in the cell type and the signal is disguised
+within the other group.
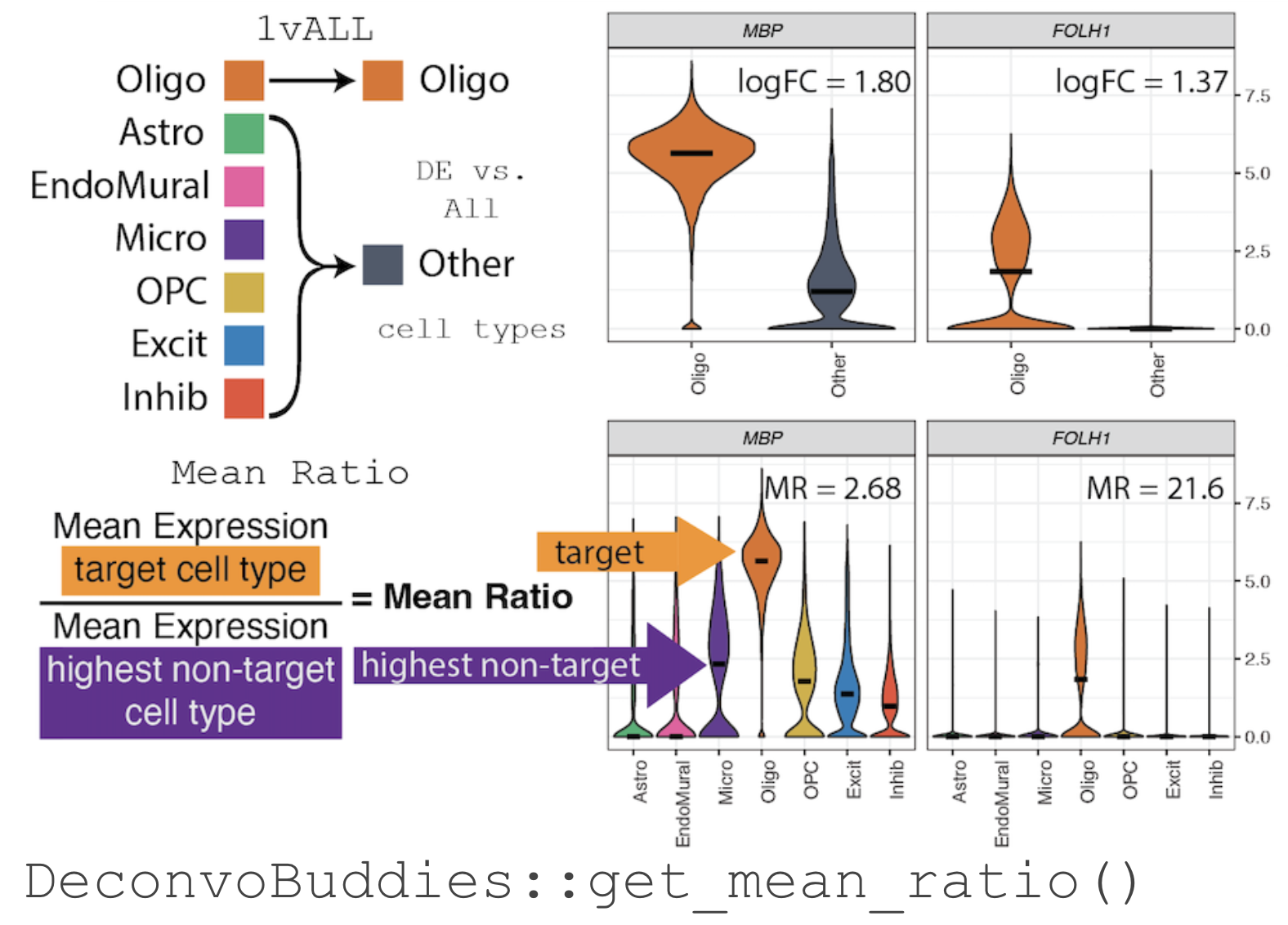
+For example, in our snRNA-seq dataset from Human DLPFC selecting
+marker gene for the cell type Oligodendrocyte (Oligo), MBP has
+a high log fold change when testing by 1vALL (see illustration
+below). But, when the expression of MBP is observed by individual cell
+types there is also expression in the related cell types Microglia
+(Micro) and Oligodendrocyte precursor cells (OPC).
+
+ +1vALL Marker Gene differential
+expression vs. MeanRatio method for marker gene selection
+
+
-
+1vALL Marker Gene differential
+expression vs. MeanRatio method for marker gene selection
+
+
-The MeanRatio Method
+The Mean Ratio Method
+
To capture genes with more cell type specific expression and less
+noise, we developed the Mean Ratio method. The
+Mean Ratio method works by selecting genes with large
+differences between gene expression in the target cell type and the
+closest non-target cell type, by evaluating genes by their
+MeanRatio metric.
+We calculate the MeanRatio for a target cell type for
+each gene by dividing the mean expression of the target cell by
+the mean expression of the next highest non-target cell type.
+Genes with the highest MeanRatio values are selected as
+marker genes.
+In the above example, Oligo is the target cell type.
+Micro has the highest mean expression out of the other non-target (not
+Oligo) cell types. The
+MeanRatio = mean expression Oligo/mean expression Micro,
+for MBP MeanRatio = 2.68 for gene FOLH1
+MeanRatio is much higher 21.6 showing FOLH1 is the better
+marker gene (in contrast to ranking by 1vALL log FC). In the
+violin plots you can see that expression of FOLH1 is much more
+specific to Oligo than MBP, supporting the ranking by
+MeanRatio.
+We have implemented the Mean Ratio method
+in this R package with the function get_mean_ratio() this
+Vignette will cover our process for marker gene selection.
Goals of this Vignette
We will be demonstrating how to use DeconvoBuddies tools
when finding cell type marker genes in single cell RNA-seq data via the
-MeanRatio method.
+MeanRatio method.
+# library("tidyr")
+# library("tibble")
+# library("ggplot2")
2. Download DLPFC snRNA-seq data.
+Here we will download single nucleus RNA-seq data from the Human
+DLPFC with 77k nuclei x 36k genes (TODO cite). This data is stored in a
+SingleCellExperiment object. The nuclei in this dataset are
+labled by cell types at a few resolutions, we will focus on the “broad”
+resolution that contains seven cell types.
## Use spatialLIBD to fetch the snRNA-seq dataset
sce_path_zip <- fetch_deconvo_data("sce")
-#> 2024-08-07 17:35:26.553214 loading file /github/home/.cache/R/BiocFileCache/4a1415db437_sce_DLPFC_annotated.zip%3Fdl%3D1
+#> 2024-08-12 15:10:22.281689 loading file /github/home/.cache/R/BiocFileCache/146b2a8cb8a7_sce_DLPFC_annotated.zip%3Fdl%3D1
## unzip and load the data
sce_path <- unzip(sce_path_zip, exdir = tempdir())
@@ -201,8 +258,6 @@ 2. Download DLPFC snRNA-seq data.sce <- sce[, sce$cellType_broad_hc != "Ambiguous"]
sce$cellType_broad_hc <- droplevels(sce$cellType_broad_hc)
-dim(sce)
-#> [1] 36601 56447
## Check the broad cell type distribution
table(sce$cellType_broad_hc)
#>
@@ -210,7 +265,200 @@ 2. Download DLPFC snRNA-seq data.#> 3979 2157 1601 10894 1940 24809 11067
## We're going to subset to the first 5k genes to save memory
-sce <- sce[seq_len(5000), ]
+## In a real application you'll want to use the full dataset
+sce <- sce[seq_len(5000), ]
+
+## check the final dimensions of the dataset
+dim(sce)
+#> [1] 5000 56447
+
+
+3. Find MeanRatio marker genes
+
+To find Mean Ratio marker genes for the data in
+sce we’ll use the function
+DeconvoBuddies::get_mean_ratio(), this function takes a
+SingleCellExperiment object sce the name of
+the column in the colData(sce) that contains the cell type
+annotations of interest (here we’ll use cellType_broad_hc),
+and optionally you can also supply additional column names from the
+rowData(sce) to add the gene_name and/or
+gene_ensembl information to the table output of
+get_mean_ratio.
+
+# calculate the Mean Ratio of genes for each cell type in sce
+mean_ratio_marker_stats <- get_mean_ratio(
+ sce = sce, # sce is the SingleCellExperiment with our data
+ assay_name = "logcounts", ## assay to use, we recommend logcounts [default]
+ cellType_col = "cellType_broad_hc", # column in colData with cell type info
+ gene_ensembl = "gene_id", # column in rowData with ensembl gene ids
+ gene_name = "gene_name" # column in rowData with gene names/symbols
+)
+The function get_mean_ratio() returns a
+tibble with the following columns:
+
+-
+
gene is the name of the gene (from
+rownames(sce)).
+-
+
cellType.target is the cell type we’re finding marker
+genes for.
+-
+
mean.target is the mean expression of gene
+for cellType.target.
+-
+
cellType.2nd is the second highest non-target cell
+type.
+-
+
mean.2nd is the mean expression of gene
+for cellType.2nd.
+-
+
MeanRatio is the ratio of
+mean.target/mean.2nd.
+-
+
MeanRatio.rank is the rank of MeanRatio
+for the cell type.
+-
+
MeanRatio.anno is an annotation of the
+MeanRatio calculation helpful for plotting.
+-
+
gene_ensembl & gene_name optional cols
+from rowData(sce) specified by the user to add gene
+information
+
+
+## Explore the tibble output
+mean_ratio_marker_stats
+#> # A tibble: 1,721 × 10
+#> gene cellType.target mean.target cellType.2nd mean.2nd MeanRatio
+#> <chr> <fct> <dbl> <fct> <dbl> <dbl>
+#> 1 LYPD6B Inhib 1.20 Excit 0.0967 12.4
+#> 2 GREM2 Inhib 1.20 Excit 0.271 4.44
+#> 3 IGSF3 Inhib 0.899 Excit 0.241 3.72
+#> 4 GALNT14 Inhib 1.86 Excit 0.503 3.69
+#> 5 LYPD6 Inhib 1.09 Astro 0.435 2.51
+#> 6 SLC35D1 Inhib 0.781 OPC 0.375 2.08
+#> 7 FLVCR1 Inhib 0.576 Excit 0.336 1.72
+#> 8 RAVER2 Inhib 1.09 OPC 0.651 1.68
+#> 9 ARHGEF11 Inhib 1.81 Excit 1.10 1.64
+#> 10 VAV3 Inhib 1.68 Astro 1.03 1.63
+#> # ℹ 1,711 more rows
+#> # ℹ 4 more variables: MeanRatio.rank <int>, MeanRatio.anno <chr>,
+#> # gene_ensembl <chr>, gene_name <chr>
+
+## genes with the highest MeanRatio are the best marker genes for each cell type
+mean_ratio_marker_stats |>
+ filter(MeanRatio.rank == 1)
+#> # A tibble: 7 × 10
+#> gene cellType.target mean.target cellType.2nd mean.2nd MeanRatio
+#> <chr> <fct> <dbl> <fct> <dbl> <dbl>
+#> 1 LYPD6B Inhib 1.20 Excit 0.0967 12.4
+#> 2 AC012494.1 Oligo 2.37 OPC 0.147 16.1
+#> 3 MIR3681HG OPC 1.58 Excit 0.205 7.69
+#> 4 AC011995.2 Excit 1.01 Inhib 0.135 7.51
+#> 5 PRDM16 Astro 1.97 EndoMural 0.142 13.9
+#> 6 SLC2A1 EndoMural 1.49 Astro 0.146 10.2
+#> 7 LINC01141 Micro 1.57 Excit 0.0640 24.5
+#> # ℹ 4 more variables: MeanRatio.rank <int>, MeanRatio.anno <chr>,
+#> # gene_ensembl <chr>, gene_name <chr>
+
+
+4. Find 1vALL marker genes
+
+To further explore cell type marker genes it can be helpful to also
+calculate the 1vALL stats for the dataset. To help with this we have
+included the function DeconvoBuddies::findMarkers_1vALL,
+which is a wrapper for scran::findMarkers() that iterates
+through cell types and creates an table output in a compatible with the
+output get_mean_ratio().
+Similarity
+Note this function can take a bit of time to run.
+
+
+marker_stats_1vAll <- findMarkers_1vAll(
+ sce = sce, # sce is the SingleCellExperiment with our data
+ assay_name = "counts",
+ cellType_col = "cellType_broad_hc", # column in colData with cell type info
+ mod = "~BrNum" # Control for donor stored in "BrNum" with mod
+)
+#> 2024-08-12 15:11:00.718289 - Find markers for: Inhib
+#> 2024-08-12 15:12:35.089049 - Find markers for: Oligo
+#> 2024-08-12 15:14:09.079623 - Find markers for: OPC
+#> 2024-08-12 15:15:43.553514 - Find markers for: Excit
+#> 2024-08-12 15:17:17.52289 - Find markers for: Astro
+#> 2024-08-12 15:18:52.162451 - Find markers for: EndoMural
+#> 2024-08-12 15:20:26.036891 - Find markers for: Micro
+#> Building Table - 2024-08-12 15:21:59.434288
+#> ** Done! **
+The function findMarkers_1vALL() returns a
+tibble with the following columns:
+
+-
+
gene is the name of the gene (from
+rownames(sce)).
+-
+
logFC the log fold change from the DE test
+-
+
log.p.value the log of the p-value of the DE test
+-
+
log.FDR the log of the False Discovery Rate adjusted
+p.value
+-
+
std.logFC the standard logFC
+-
+
cellType.target the cell type we’re finding marker
+genes for
+-
+
std.logFC.rank the rank of std.logFC for
+each cell type
+-
+
std.logFC.anno is an annotation of the
+std.logFC value helpful for plotting.
+
+
+## Explore the tibble output
+marker_stats_1vAll
+#> # A tibble: 35,000 × 8
+#> # Groups: cellType.target [7]
+#> gene logFC log.p.value log.FDR std.logFC cellType.target std.logFC.rank
+#> <chr> <dbl> <dbl> <dbl> <dbl> <fct> <int>
+#> 1 SILC1 2.77 -6747. -6739. 1.42 Inhib 1
+#> 2 LYPD6B 2.48 -6338. -6330. 1.37 Inhib 2
+#> 3 LYPD6 2.18 -6147. -6140. 1.35 Inhib 3
+#> 4 ALK 4.56 -5707. -5700. 1.30 Inhib 4
+#> 5 WLS 3.26 -5111. -5104. 1.22 Inhib 5
+#> 6 PLD5 5.85 -4711. -4704. 1.17 Inhib 6
+#> 7 KIRREL1 1.04 -4539. -4532. 1.14 Inhib 7
+#> 8 TRIM67 0.622 -4326. -4319. 1.11 Inhib 8
+#> 9 GREM2 2.11 -4048. -4042. 1.07 Inhib 9
+#> 10 IGSF3 1.27 -3783. -3777. 1.04 Inhib 10
+#> # ℹ 34,990 more rows
+#> # ℹ 1 more variable: std.logFC.anno <chr>
+
+## genes with the highest MeanRatio are the best marker genes for each cell type
+marker_stats_1vAll |>
+ filter(std.logFC.rank == 1)
+#> # A tibble: 7 × 8
+#> # Groups: cellType.target [7]
+#> gene logFC log.p.value log.FDR std.logFC cellType.target std.logFC.rank
+#> <chr> <dbl> <dbl> <dbl> <dbl> <fct> <int>
+#> 1 SILC1 2.77 -6747. -6739. 1.42 Inhib 1
+#> 2 RNF220 10.7 -8373. -8365. 1.52 Oligo 1
+#> 3 BX284613 7.42 -12763. -12754. 4.20 OPC 1
+#> 4 KIAA1211L 7.89 -8371. -8362. 1.23 Excit 1
+#> 5 PRDM16 1.90 -8853. -8844. 2.38 Astro 1
+#> 6 EPAS1 6.50 -9480. -9471. 3.31 EndoMural 1
+#> 7 CSF3R 0.808 -9670. -9661. 3.86 Micro 1
+#> # ℹ 1 more variable: std.logFC.anno <chr>
+
+
+
Reproducibility
@@ -242,7 +490,7 @@ Reproducibility
This package was developed using biocthis.
Code for creating the vignette
-
+
## Create the vignette
library("rmarkdown")
system.time(render("Marker_Finding.Rmd", "BiocStyle::html_document"))
@@ -251,9 +499,9 @@ Reproducibilitylibrary("knitr")
knit("Marker_Finding.Rmd", tangle = TRUE)
Date the vignette was generated.
-#> [1] "2024-08-07 17:35:41 UTC"
+#> [1] "2024-08-12 15:21:59 UTC"
Wallclock time spent generating the vignette.
-#> Time difference of 27.362 secs
+#> Time difference of 11.842 mins
R session information.
#> ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
#> setting value
@@ -265,8 +513,8 @@ Reproducibility#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz UTC
-#> date 2024-08-07
-#> pandoc 3.2 @ /usr/bin/ (via rmarkdown)
+#> date 2024-08-12
+#> pandoc 3.3 @ /usr/bin/ (via rmarkdown)
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
@@ -310,7 +558,7 @@ Reproducibility#> data.table 1.15.4 2024-03-30 [1] RSPM (R 4.4.0)
#> DBI 1.2.3 2024-06-02 [1] RSPM (R 4.4.0)
#> dbplyr 2.5.0 2024-03-19 [1] RSPM (R 4.4.0)
-#> DeconvoBuddies * 0.99.0 2024-08-07 [1] Bioconductor
+#> DeconvoBuddies * 0.99.0 2024-08-12 [1] Bioconductor
#> DelayedArray 0.31.11 2024-08-04 [1] Bioconductor 3.20 (R 4.4.1)
#> desc 1.4.3 2023-12-10 [2] RSPM (R 4.4.0)
#> digest 0.6.36 2024-06-23 [2] RSPM (R 4.4.0)
@@ -334,7 +582,7 @@ Reproducibility#> GenomicAlignments 1.41.0 2024-05-01 [1] Bioconductor 3.20 (R 4.4.0)
#> GenomicRanges * 1.57.1 2024-06-12 [1] Bioconductor 3.20 (R 4.4.0)
#> ggbeeswarm 0.7.2 2023-04-29 [1] RSPM (R 4.4.0)
-#> ggplot2 * 3.5.1 2024-04-23 [1] RSPM (R 4.4.0)
+#> ggplot2 3.5.1 2024-04-23 [1] RSPM (R 4.4.0)
#> ggrepel 0.9.5 2024-01-10 [1] RSPM (R 4.4.0)
#> glue 1.7.0 2024-01-09 [2] RSPM (R 4.4.0)
#> golem 0.4.1 2023-06-05 [1] RSPM (R 4.4.0)
@@ -373,7 +621,7 @@ Reproducibility#> paletteer 1.6.0 2024-01-21 [1] RSPM (R 4.4.0)
#> pillar 1.9.0 2023-03-22 [2] RSPM (R 4.4.0)
#> pkgconfig 2.0.3 2019-09-22 [2] RSPM (R 4.4.0)
-#> pkgdown 2.1.0.9000 2024-08-05 [1] Github (r-lib/pkgdown@1d40a80)
+#> pkgdown 2.1.0.9000 2024-08-12 [1] Github (r-lib/pkgdown@1d40a80)
#> plotly 4.10.4 2024-01-13 [1] RSPM (R 4.4.0)
#> plyr 1.8.9 2023-10-02 [1] RSPM (R 4.4.0)
#> png 0.1-8 2022-11-29 [1] RSPM (R 4.4.0)
@@ -421,8 +669,8 @@ Reproducibility#> SummarizedExperiment * 1.35.1 2024-06-28 [1] Bioconductor 3.20 (R 4.4.1)
#> systemfonts 1.1.0 2024-05-15 [2] RSPM (R 4.4.0)
#> textshaping 0.4.0 2024-05-24 [2] RSPM (R 4.4.0)
-#> tibble * 3.2.1 2023-03-20 [2] RSPM (R 4.4.0)
-#> tidyr * 1.3.1 2024-01-24 [1] RSPM (R 4.4.0)
+#> tibble 3.2.1 2023-03-20 [2] RSPM (R 4.4.0)
+#> tidyr 1.3.1 2024-01-24 [1] RSPM (R 4.4.0)
#> tidyselect 1.2.1 2024-03-11 [1] RSPM (R 4.4.0)
#> timechange 0.3.0 2024-01-18 [1] RSPM (R 4.4.0)
#> UCSC.utils 1.1.0 2024-05-01 [1] Bioconductor 3.20 (R 4.4.0)
diff --git a/pkgdown.yml b/pkgdown.yml
index 69bd4aa..9c8847f 100644
--- a/pkgdown.yml
+++ b/pkgdown.yml
@@ -1,8 +1,8 @@
-pandoc: '3.2'
+pandoc: '3.3'
pkgdown: 2.1.0.9000
pkgdown_sha: 1d40a80e6b3564a6d7da0ce467b0a4570aa5665e
articles:
DeconvoBuddies: DeconvoBuddies.html
Deconvolution_Benchmark_DLPFC: Deconvolution_Benchmark_DLPFC.html
Marker_Finding: Marker_Finding.html
-last_built: 2024-08-07T17:32Z
+last_built: 2024-08-12T15:07Z
diff --git a/reference/dot-get_mean_ratio2.html b/reference/dot-get_mean_ratio2.html
index a2ba3ce..4b8a2dd 100644
--- a/reference/dot-get_mean_ratio2.html
+++ b/reference/dot-get_mean_ratio2.html
@@ -117,7 +117,7 @@ Details
Examples
#' ## load example SingleCellExperiment
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:32:53.195728 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:07:39.587022 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## Get the mean ratio for each gene for each cell type defined in `cellType_broad_hc`
.get_mean_ratio2(sce_DLPFC_example, cellType_col = "cellType_broad_hc")
diff --git a/reference/fetch_deconvo_data.html b/reference/fetch_deconvo_data.html
index 9eb8fbc..806519d 100644
--- a/reference/fetch_deconvo_data.html
+++ b/reference/fetch_deconvo_data.html
@@ -132,9 +132,9 @@ Examples
## A RangedSummarizedExperiment (41.16 MB)
if (!exists("rse-gene")) rse_gene <- fetch_deconvo_data("rse_gene")
-#> 2024-08-07 17:32:55.846502 loading file /github/home/.cache/R/BiocFileCache/4a1de5efa9_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
+#> 2024-08-12 15:07:42.170264 loading file /github/home/.cache/R/BiocFileCache/146b2dbc692f_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
-## explore data
+## explore bulk data
rse_gene
#> class: RangedSummarizedExperiment
#> dim: 21745 110
@@ -147,22 +147,13 @@ Examples
#> 2107UNHS-0291_Br2720_Mid_Cyto ... AN00000906_Br8667_Mid_Cyto
#> AN00000906_Br8667_Mid_Nuc
#> colData names(80): SAMPLE_ID Sample ... diagnosis qc_class
-# class: RangedSummarizedExperiment
-# dim: 21745 110
-# metadata(1): SPEAQeasy_settings
-# assays(2): counts logcounts
-# rownames(21745): ENSG00000227232.5 ENSG00000278267.1 ... ENSG00000210195.2 ENSG00000210196.2
-# rowData names(11): Length gencodeID ... gencodeTx passExprsCut
-# colnames(110): 2107UNHS-0291_Br2720_Mid_Bulk 2107UNHS-0291_Br2720_Mid_Cyto ... AN00000906_Br8667_Mid_Cyto
-# AN00000906_Br8667_Mid_Nuc
-# colData names(78): SAMPLE_ID Sample ... diagnosis qc_class
## load example snRNA-seq data
## A SingleCellExperiment (4.79 MB)
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:32:57.693436 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
-
+#> 2024-08-12 15:07:43.289595 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+## explore example sce data
sce_DLPFC_example
#> class: SingleCellExperiment
#> dim: 557 10000
@@ -176,17 +167,6 @@ Examples
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
-# class: SingleCellExperiment
-# dim: 557 10000
-# metadata(3): Samples cell_type_colors cell_type_colors_broad
-# assays(2): counts logcounts
-# rownames(557): GABRD PRDM16 ... AFF2 MAMLD1
-# rowData names(7): source type ... gene_type binomial_deviance
-# colnames(10000): 16_CGAAGTTTCGCACGAC-1 11_TTGGGATCAACCGCTG-1 ... 8_CGCATAAGTTAAACCC-1 16_AGCTACATCCCGAGAC-1
-# colData names(32): Sample Barcode ... cellType_layer layer_annotation
-# reducedDimNames(0):
-# mainExpName: NULL
-# altExpNames(0):
## check the logcounts
SingleCellExperiment::logcounts(sce_DLPFC_example)[1:5, 1:5]
@@ -204,6 +184,7 @@ Examples
#> ADGRB2 2.253454 0.0000000
if (FALSE) { # \dontrun{
+## download the full sce experiment object
sce_path_zip <- fetch_deconvo_data("sce")
sce_path <- unzip(sce_path_zip, exdir = tempdir())
sce <- HDF5Array::loadHDF5SummarizedExperiment(
diff --git a/reference/findMarkers_1vAll.html b/reference/findMarkers_1vAll.html
index cb26561..14cec90 100644
--- a/reference/findMarkers_1vAll.html
+++ b/reference/findMarkers_1vAll.html
@@ -76,7 +76,7 @@ Calculate 1 vs. All standard fold change for each gene x cell type, wrapper
assay_name = "counts",
cellType_col = "cellType",
add_symbol = FALSE,
- mod = "~donor",
+ mod = NULL,
verbose = TRUE
)
@@ -102,7 +102,9 @@ Arguments
mod
-String specifying the model used as design in findMarkers. Can be NULL if there are no blocking terms with uninteresting factors as documented at pairwiseTTests.
String specifying the model used as design in findMarkers.
+Can be NULL (default) if there are no blocking terms with uninteresting
+factors as documented at pairwiseTTests.
verbose
@@ -111,28 +113,88 @@ Arguments
Value
- Table of 1 vs. ALL std log fold change + p-values for each gene x cell type
-
+ Tibble of 1 vs. ALL std log fold change + p-values for each gene x cell type
gene is the name of the gene (from rownames(sce)).
logFC the log fold change from the DE test
log.p.value the log of the p-value of the DE test
log.FDR the log of the False Discovery Rate adjusted p.value
std.logFC the standard logFC
cellType.target the cell type we're finding marker genes for
std.logFC.rank the rank of std.logFC for each cell type
std.logFC.anno is an annotation of the std.logFC value
+helpful for plotting.
Examples
- markers_1vAll <- findMarkers_1vAll(sce_ab)
-#> A - '2024-08-07 17:32:58.690142
-#> B - '2024-08-07 17:32:59.008849
-#> Building Table - 2024-08-07 17:32:59.164719
+ ## load example SingleCellExperiment
+if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
+#> 2024-08-12 15:07:45.774661 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+## Explore properties of the sce object
+sce_DLPFC_example
+#> class: SingleCellExperiment
+#> dim: 557 10000
+#> metadata(3): Samples cell_type_colors cell_type_colors_broad
+#> assays(1): logcounts
+#> rownames(557): GABRD PRDM16 ... AFF2 MAMLD1
+#> rowData names(7): source type ... gene_type binomial_deviance
+#> colnames(10000): 8_AGTGACTGTAGTTACC-1 17_GCAGCCAGTGAGTCAG-1 ...
+#> 12_GGACGTCTCTGACAGT-1 1_GGTTAACTCTCTCTAA-1
+#> colData names(32): Sample Barcode ... cellType_layer layer_annotation
+#> reducedDimNames(0):
+#> mainExpName: NULL
+#> altExpNames(0):
+
+## this data contains logcounts of gene expression
+SummarizedExperiment::assays(sce_DLPFC_example)$logcounts[1:5, 1:5]
+#> 8_AGTGACTGTAGTTACC-1 17_GCAGCCAGTGAGTCAG-1 3_CTGGACGAGCTTCATG-1
+#> GABRD 0 0.9249246 0.000000
+#> PRDM16 0 0.0000000 0.000000
+#> MICOS10 0 0.0000000 0.000000
+#> LINC01141 0 0.0000000 0.000000
+#> ADGRB2 0 0.9249246 2.253612
+#> 13_CCCTCAAAGTCTAGCT-1 11_TGTAAGCCATTCTGTT-1
+#> GABRD 0.000000 0.0000000
+#> PRDM16 0.000000 0.0000000
+#> MICOS10 0.000000 0.6528615
+#> LINC01141 0.000000 0.0000000
+#> ADGRB2 2.253454 0.0000000
+
+## nuclei are classified in to cell types
+table(sce_DLPFC_example$cellType_broad_hc)
+#>
+#> Astro EndoMural Micro Oligo OPC Excit Inhib
+#> 692 417 316 1970 350 4335 1920
+
+## Get the 1vALL stats for each gene for each cell type defined in `cellType_broad_hc`
+marker_stats_1vAll <- findMarkers_1vAll(
+ sce = sce_DLPFC_example,
+ assay_name = "logcounts",
+ cellType_col = "cellType_broad_hc",
+ mod = "~BrNum"
+)
+#> 2024-08-12 15:07:46.02474 - Find markers for: Oligo
+#> 2024-08-12 15:07:46.488802 - Find markers for: Excit
+#> 2024-08-12 15:07:46.874934 - Find markers for: Inhib
+#> 2024-08-12 15:07:47.995957 - Find markers for: Micro
+#> 2024-08-12 15:07:48.331533 - Find markers for: OPC
+#> 2024-08-12 15:07:48.667361 - Find markers for: EndoMural
+#> 2024-08-12 15:07:49.036291 - Find markers for: Astro
+#> Building Table - 2024-08-12 15:07:49.415034
#> ** Done! **
-head(markers_1vAll)
+
+## explore output, top markers have high logFC
+head(marker_stats_1vAll)
#> # A tibble: 6 × 8
-#> # Groups: cellType.target [2]
-#> gene logFC log.p.value log.FDR std.logFC cellType.target rank_marker
-#> <chr> <dbl> <dbl> <dbl> <dbl> <chr> <int>
-#> 1 G-D1_A 0.5 -3.65e+ 1 -3.58e+ 1 1.97 A 1
-#> 2 G-D2_A 0.5 -3.65e+ 1 -3.58e+ 1 1.97 A 2
-#> 3 G-D1_B -0.5 -1.42e-16 -1.42e-16 -1.97 A 3
-#> 4 G-D2_B -0.5 -1.42e-16 -1.42e-16 -1.97 A 4
-#> 5 G-D2_B 0.5 -3.65e+ 1 -3.58e+ 1 1.97 B 1
-#> 6 G-D1_B 0.5 -3.65e+ 1 -3.58e+ 1 1.97 B 2
-#> # ℹ 1 more variable: anno_logFC <chr>
+#> # Groups: cellType.target [1]
+#> gene logFC log.p.value log.FDR std.logFC cellType.target std.logFC.rank
+#> <chr> <dbl> <dbl> <dbl> <dbl> <fct> <int>
+#> 1 ST18 4.46 -6697. -6691. 4.32 Oligo 1
+#> 2 PLP1 4.25 -5233. -5227. 3.50 Oligo 2
+#> 3 MOBP 3.18 -4821. -4816. 3.28 Oligo 3
+#> 4 ENPP2 2.72 -4521. -4516. 3.12 Oligo 4
+#> 5 TF 3.00 -4466. -4462. 3.09 Oligo 5
+#> 6 RNF220 3.80 -4390. -4385. 3.05 Oligo 6
+#> # ℹ 1 more variable: std.logFC.anno <chr>
diff --git a/reference/get_mean_ratio.html b/reference/get_mean_ratio.html
index e96dc51..c99630d 100644
--- a/reference/get_mean_ratio.html
+++ b/reference/get_mean_ratio.html
@@ -121,8 +121,10 @@ Value
mean.2nd is the mean expression of gene for cellType.2nd.
MeanRatio is the ratio of mean.target/mean.2nd.
MeanRatio.rank is the rank of MeanRatio for the cell type.
MeanRatio.anno is an annotation of the MeanRatio calculation helpful for plotting.
gene_ensembl & gene_name optional cols spcified by the user to add gene infomation
MeanRatio.anno is an annotation of the MeanRatio calculation helpful
+for plotting.
gene_ensembl & gene_name optional cols from rowData(sce)specified by
+the user to add gene information
Details
@@ -134,7 +136,7 @@ Details
Examples
## load example SingleCellExperiment
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:00.419412 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:07:50.863967 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## Explore properties of the sce object
sce_DLPFC_example
#> class: SingleCellExperiment
@@ -221,7 +223,10 @@ Examples
#> MAMLD1 protein_coding 75492.1
## specify rowData col names for gene_name and gene_ensembl
-get_mean_ratio(sce_DLPFC_example, cellType_col = "cellType_broad_hc", gene_name = "gene_name", gene_ensembl = "gene_id")
+ get_mean_ratio(sce_DLPFC_example,
+ cellType_col = "cellType_broad_hc",
+ gene_name = "gene_name",
+ gene_ensembl = "gene_id")
#> # A tibble: 762 × 10
#> gene cellType.target mean.target cellType.2nd mean.2nd MeanRatio
#> <chr> <fct> <dbl> <fct> <dbl> <dbl>
diff --git a/reference/plot_gene_express.html b/reference/plot_gene_express.html
index 413056c..1b3d700 100644
--- a/reference/plot_gene_express.html
+++ b/reference/plot_gene_express.html
@@ -144,7 +144,7 @@ Examples
 if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:06.529501 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:07:56.587053 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## plot expression of two genes
plot_gene_express(sce = sce_DLPFC_example, category = "cellType_broad_hc", genes = c("GAD2", "CD22"))
#> No summary function supplied, defaulting to `mean_se()`
diff --git a/reference/plot_marker_express.html b/reference/plot_marker_express.html
index 9dc7169..dc08ee0 100644
--- a/reference/plot_marker_express.html
+++ b/reference/plot_marker_express.html
@@ -155,7 +155,7 @@
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:06.529501 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:07:56.587053 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## plot expression of two genes
plot_gene_express(sce = sce_DLPFC_example, category = "cellType_broad_hc", genes = c("GAD2", "CD22"))
#> No summary function supplied, defaulting to `mean_se()`
diff --git a/reference/plot_marker_express.html b/reference/plot_marker_express.html
index 9dc7169..dc08ee0 100644
--- a/reference/plot_marker_express.html
+++ b/reference/plot_marker_express.html
@@ -155,7 +155,7 @@ See also
Examples
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:09.904147 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:07:59.949517 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## plot the top markers for Astrocytes in
plot_marker_express(
sce = sce_DLPFC_example,
diff --git a/reference/plot_marker_express_ALL.html b/reference/plot_marker_express_ALL.html
index 6749dc8..1e360ad 100644
--- a/reference/plot_marker_express_ALL.html
+++ b/reference/plot_marker_express_ALL.html
@@ -153,7 +153,7 @@ See also
Examples
#' ## Fetch sce example data
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:12.916615 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:08:02.934026 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
# Plot marker gene expression to PDF, one page per cell type in stats
pdf_file <- tempfile("test_marker_expression_ALL", fileext = ".pdf")
diff --git a/reference/plot_marker_express_List.html b/reference/plot_marker_express_List.html
index afdac08..1ce1204 100644
--- a/reference/plot_marker_express_List.html
+++ b/reference/plot_marker_express_List.html
@@ -134,7 +134,7 @@ See also
Examples
## Fetch sce example data
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:29.315545 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:08:19.448584 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## Create list-of-lists of genes to plot, names of sub-list become title of page
my_gene_list <- list(Inhib = c("GAD2", "SAMD5"), Astro = c("RGS20", "PRDM16"))
## Access and snRNA-seq example data
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:45.703235 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:08:35.815188 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## Explore snRNA-seq data in sce_DLPFC_example
sce_DLPFC_example
@@ -239,7 +239,7 @@ Access Data## Access Bulk RNA-seq data
if (!exists("rse_gene")) rse_gene <- fetch_deconvo_data("rse_gene")
-#> 2024-08-07 17:33:47.478342 loading file /github/home/.cache/R/BiocFileCache/4a1de5efa9_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
+#> 2024-08-12 15:08:37.663146 loading file /github/home/.cache/R/BiocFileCache/146b2dbc692f_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
## Explore bulk data in rse_gene
rse_gene
@@ -470,9 +470,9 @@ Reproducibilitylibrary("knitr")
knit("DeconvoBuddies.Rmd", tangle = TRUE)
#> [1] "2024-08-07 17:33:51 UTC"#> [1] "2024-08-12 15:08:43 UTC"#> Time difference of 18.582 secs#> Time difference of 19.447 secsR session information.#> ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
#> setting value
@@ -484,8 +484,8 @@ Reproducibility#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz UTC
-#> date 2024-08-07
-#> pandoc 3.2 @ /usr/bin/ (via rmarkdown)
+#> date 2024-08-12
+#> pandoc 3.3 @ /usr/bin/ (via rmarkdown)
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
@@ -529,7 +529,7 @@ Reproducibility#> data.table 1.15.4 2024-03-30 [1] RSPM (R 4.4.0)
#> DBI 1.2.3 2024-06-02 [1] RSPM (R 4.4.0)
#> dbplyr 2.5.0 2024-03-19 [1] RSPM (R 4.4.0)
-#> DeconvoBuddies * 0.99.0 2024-08-07 [1] Bioconductor
+#> DeconvoBuddies * 0.99.0 2024-08-12 [1] Bioconductor
#> DelayedArray 0.31.11 2024-08-04 [1] Bioconductor 3.20 (R 4.4.1)
#> desc 1.4.3 2023-12-10 [2] RSPM (R 4.4.0)
#> digest 0.6.36 2024-06-23 [2] RSPM (R 4.4.0)
@@ -594,7 +594,7 @@ Reproducibility#> paletteer 1.6.0 2024-01-21 [1] RSPM (R 4.4.0)
#> pillar 1.9.0 2023-03-22 [2] RSPM (R 4.4.0)
#> pkgconfig 2.0.3 2019-09-22 [2] RSPM (R 4.4.0)
-#> pkgdown 2.1.0.9000 2024-08-05 [1] Github (r-lib/pkgdown@1d40a80)
+#> pkgdown 2.1.0.9000 2024-08-12 [1] Github (r-lib/pkgdown@1d40a80)
#> plotly 4.10.4 2024-01-13 [1] RSPM (R 4.4.0)
#> plyr 1.8.9 2023-10-02 [1] RSPM (R 4.4.0)
#> png 0.1-8 2022-11-29 [1] RSPM (R 4.4.0)
diff --git a/articles/Deconvolution_Benchmark_DLPFC.html b/articles/Deconvolution_Benchmark_DLPFC.html
index b842b41..d83f1e8 100644
--- a/articles/Deconvolution_Benchmark_DLPFC.html
+++ b/articles/Deconvolution_Benchmark_DLPFC.html
@@ -98,7 +98,7 @@ Louise
Campus
lahuuki@gmail.com
- 7 August 2024
+ 12 August 2024
Source: vignettes/Deconvolution_Benchmark_DLPFC.Rmd
Deconvolution_Benchmark_DLPFC.Rmd
@@ -138,46 +138,46 @@ Deconvolution Methods
+
Approach
Method
Citation
Availability
-
+
weighted least squares
DWLS
Tsoucas et al, Nature Comm, 2019
R
Package Cran
-
+
Bias correction: Assay
Bisque
Jew et al, Nature Comm, 2020
R Package github
-
+
Bias correction: Sourse
MuSiC
Wang et al, Nature Communications, 2019
R Package github
-
+
Machine Learning
CIBERSORTx
Newman et al., Nature BioTech, 2019
Webtool
-
+
Bayesian
BayesPrism
Chu et al., Nature Cancer, 2022
Webtool/R
Package
-
+
linear
Hspe
Hunt et al., Ann. Appl. Stat, 2021
@@ -261,7 +261,7 @@ Bulk RNA-seq data
## use fetch deconvon data to load rse_gene
rse_gene <- fetch_deconvo_data("rse_gene")
-#> 2024-08-07 17:34:07.847847 loading file /github/home/.cache/R/BiocFileCache/4a1de5efa9_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
+#> 2024-08-12 15:08:59.333378 loading file /github/home/.cache/R/BiocFileCache/146b2dbc692f_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
rse_gene
#> class: RangedSummarizedExperiment
#> dim: 21745 110
@@ -297,7 +297,7 @@ Reference snRNA-seq data
## Use spatialLIBD to fetch the snRNA-seq dataset
sce_path_zip <- fetch_deconvo_data("sce")
-#> 2024-08-07 17:34:09.528637 loading file /github/home/.cache/R/BiocFileCache/4a1415db437_sce_DLPFC_annotated.zip%3Fdl%3D1
+#> 2024-08-12 15:09:01.898358 loading file /github/home/.cache/R/BiocFileCache/146b2a8cb8a7_sce_DLPFC_annotated.zip%3Fdl%3D1
## unzip and load the data
sce_path <- unzip(sce_path_zip, exdir = tempdir())
@@ -388,10 +388,12 @@ 3. Select Marker Genes
We have developed a method for finding marker genes called the “Mean
-Ratio”. We calculate the Mean Ratio for a target cell type
+Ratio”. We calculate the MeanRatio for a target cell type
for each gene by dividing the mean expression of the target cell by the
mean expression of the next highest non-target cell type. Genes with the
-highest Mean Ratio values are selected as marker genes.
+highest MeanRatio values are selected as marker genes.
+For a tutorial on marker gene selection check out Vignette:
+Deconvolution Benchmark in Human DLPFC.
@@ -762,9 +764,9 @@ Reproducibilitylibrary("knitr")
knit("Deconvolution_Benchmark_DLPFC.Rmd", tangle = TRUE)
Date the vignette was generated.
-#> [1] "2024-08-07 17:35:08 UTC"
+#> [1] "2024-08-12 15:10:03 UTC"
Wallclock time spent generating the vignette.
-#> Time difference of 1.214 mins
+#> Time difference of 1.27 mins
R session information.
#> ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
#> setting value
@@ -776,8 +778,8 @@ Reproducibility#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz UTC
-#> date 2024-08-07
-#> pandoc 3.2 @ /usr/bin/ (via rmarkdown)
+#> date 2024-08-12
+#> pandoc 3.3 @ /usr/bin/ (via rmarkdown)
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
@@ -822,7 +824,7 @@ Reproducibility#> data.table 1.15.4 2024-03-30 [1] RSPM (R 4.4.0)
#> DBI 1.2.3 2024-06-02 [1] RSPM (R 4.4.0)
#> dbplyr 2.5.0 2024-03-19 [1] RSPM (R 4.4.0)
-#> DeconvoBuddies * 0.99.0 2024-08-07 [1] Bioconductor
+#> DeconvoBuddies * 0.99.0 2024-08-12 [1] Bioconductor
#> DelayedArray 0.31.11 2024-08-04 [1] Bioconductor 3.20 (R 4.4.1)
#> desc 1.4.3 2023-12-10 [2] RSPM (R 4.4.0)
#> digest 0.6.36 2024-06-23 [2] RSPM (R 4.4.0)
@@ -891,7 +893,7 @@ Reproducibility#> paletteer 1.6.0 2024-01-21 [1] RSPM (R 4.4.0)
#> pillar 1.9.0 2023-03-22 [2] RSPM (R 4.4.0)
#> pkgconfig 2.0.3 2019-09-22 [2] RSPM (R 4.4.0)
-#> pkgdown 2.1.0.9000 2024-08-05 [1] Github (r-lib/pkgdown@1d40a80)
+#> pkgdown 2.1.0.9000 2024-08-12 [1] Github (r-lib/pkgdown@1d40a80)
#> plotly 4.10.4 2024-01-13 [1] RSPM (R 4.4.0)
#> plyr 1.8.9 2023-10-02 [1] RSPM (R 4.4.0)
#> png 0.1-8 2022-11-29 [1] RSPM (R 4.4.0)
diff --git a/articles/Marker_Finding.html b/articles/Marker_Finding.html
index 54c0154..8626235 100644
--- a/articles/Marker_Finding.html
+++ b/articles/Marker_Finding.html
@@ -98,7 +98,7 @@ Louise
Campus
lahuuki@gmail.com
- 7 August 2024
+ 12 August 2024
Source: vignettes/Marker_Finding.Rmd
Marker_Finding.Rmd
@@ -113,21 +113,74 @@ Introduction
What are Marker Genes?
+Cell type marker genes have cell type specific expression, that is
+high expression in the target cell type, and low expression in all other
+cell types. Sub-setting the genes considered in a cell type
+deconvolution analysis helps reduce noise and can improve the accuracy
+of a deconvolution method.
How can we select marker genes?
+There are several approaches to select marker genes.
+One popular method is “1 vs. All” differential expression (Lun et
+al., 2016, F1000Res) (TODO update citation), where genes are tested for
+differential expression between the target cell type, and a combined
+group of all “other” cell types. Statistically
+significant differntially expressed genes (DEGs) can be selected as a
+set of marker genes, DEGs can be ranked by high log fold change.
+However in some cases 1vAll can select genes with high
+expression in non-target cell types, especially in cell types related to
+the target cell types (such as Neuron sub-types), or when there is a
+smaller number of cells in the cell type and the signal is disguised
+within the other group.
+For example, in our snRNA-seq dataset from Human DLPFC selecting
+marker gene for the cell type Oligodendrocyte (Oligo), MBP has
+a high log fold change when testing by 1vALL (see illustration
+below). But, when the expression of MBP is observed by individual cell
+types there is also expression in the related cell types Microglia
+(Micro) and Oligodendrocyte precursor cells (OPC).
+
+
+1vALL Marker Gene differential
+expression vs. MeanRatio method for marker gene selection
+
+
-The MeanRatio Method
+The Mean Ratio Method
+
To capture genes with more cell type specific expression and less
+noise, we developed the Mean Ratio method. The
+Mean Ratio method works by selecting genes with large
+differences between gene expression in the target cell type and the
+closest non-target cell type, by evaluating genes by their
+MeanRatio metric.
+We calculate the MeanRatio for a target cell type for
+each gene by dividing the mean expression of the target cell by
+the mean expression of the next highest non-target cell type.
+Genes with the highest MeanRatio values are selected as
+marker genes.
+In the above example, Oligo is the target cell type.
+Micro has the highest mean expression out of the other non-target (not
+Oligo) cell types. The
+MeanRatio = mean expression Oligo/mean expression Micro,
+for MBP MeanRatio = 2.68 for gene FOLH1
+MeanRatio is much higher 21.6 showing FOLH1 is the better
+marker gene (in contrast to ranking by 1vALL log FC). In the
+violin plots you can see that expression of FOLH1 is much more
+specific to Oligo than MBP, supporting the ranking by
+MeanRatio.
+We have implemented the Mean Ratio method
+in this R package with the function get_mean_ratio() this
+Vignette will cover our process for marker gene selection.
Goals of this Vignette
We will be demonstrating how to use DeconvoBuddies tools
when finding cell type marker genes in single cell RNA-seq data via the
-MeanRatio method.
+MeanRatio method.
+# library("tidyr")
+# library("tibble")
+# library("ggplot2")
2. Download DLPFC snRNA-seq data.
+Here we will download single nucleus RNA-seq data from the Human
+DLPFC with 77k nuclei x 36k genes (TODO cite). This data is stored in a
+SingleCellExperiment object. The nuclei in this dataset are
+labled by cell types at a few resolutions, we will focus on the “broad”
+resolution that contains seven cell types.
## Use spatialLIBD to fetch the snRNA-seq dataset
sce_path_zip <- fetch_deconvo_data("sce")
-#> 2024-08-07 17:35:26.553214 loading file /github/home/.cache/R/BiocFileCache/4a1415db437_sce_DLPFC_annotated.zip%3Fdl%3D1
+#> 2024-08-12 15:10:22.281689 loading file /github/home/.cache/R/BiocFileCache/146b2a8cb8a7_sce_DLPFC_annotated.zip%3Fdl%3D1
## unzip and load the data
sce_path <- unzip(sce_path_zip, exdir = tempdir())
@@ -201,8 +258,6 @@ 2. Download DLPFC snRNA-seq data.sce <- sce[, sce$cellType_broad_hc != "Ambiguous"]
sce$cellType_broad_hc <- droplevels(sce$cellType_broad_hc)
-dim(sce)
-#> [1] 36601 56447
## Check the broad cell type distribution
table(sce$cellType_broad_hc)
#>
@@ -210,7 +265,200 @@ 2. Download DLPFC snRNA-seq data.#> 3979 2157 1601 10894 1940 24809 11067
## We're going to subset to the first 5k genes to save memory
-sce <- sce[seq_len(5000), ]
+## In a real application you'll want to use the full dataset
+sce <- sce[seq_len(5000), ]
+
+## check the final dimensions of the dataset
+dim(sce)
+#> [1] 5000 56447
+
+
+3. Find MeanRatio marker genes
+
+To find Mean Ratio marker genes for the data in
+sce we’ll use the function
+DeconvoBuddies::get_mean_ratio(), this function takes a
+SingleCellExperiment object sce the name of
+the column in the colData(sce) that contains the cell type
+annotations of interest (here we’ll use cellType_broad_hc),
+and optionally you can also supply additional column names from the
+rowData(sce) to add the gene_name and/or
+gene_ensembl information to the table output of
+get_mean_ratio.
+
+# calculate the Mean Ratio of genes for each cell type in sce
+mean_ratio_marker_stats <- get_mean_ratio(
+ sce = sce, # sce is the SingleCellExperiment with our data
+ assay_name = "logcounts", ## assay to use, we recommend logcounts [default]
+ cellType_col = "cellType_broad_hc", # column in colData with cell type info
+ gene_ensembl = "gene_id", # column in rowData with ensembl gene ids
+ gene_name = "gene_name" # column in rowData with gene names/symbols
+)
+The function get_mean_ratio() returns a
+tibble with the following columns:
+
+-
+
gene is the name of the gene (from
+rownames(sce)).
+-
+
cellType.target is the cell type we’re finding marker
+genes for.
+-
+
mean.target is the mean expression of gene
+for cellType.target.
+-
+
cellType.2nd is the second highest non-target cell
+type.
+-
+
mean.2nd is the mean expression of gene
+for cellType.2nd.
+-
+
MeanRatio is the ratio of
+mean.target/mean.2nd.
+-
+
MeanRatio.rank is the rank of MeanRatio
+for the cell type.
+-
+
MeanRatio.anno is an annotation of the
+MeanRatio calculation helpful for plotting.
+-
+
gene_ensembl & gene_name optional cols
+from rowData(sce) specified by the user to add gene
+information
+
+
+## Explore the tibble output
+mean_ratio_marker_stats
+#> # A tibble: 1,721 × 10
+#> gene cellType.target mean.target cellType.2nd mean.2nd MeanRatio
+#> <chr> <fct> <dbl> <fct> <dbl> <dbl>
+#> 1 LYPD6B Inhib 1.20 Excit 0.0967 12.4
+#> 2 GREM2 Inhib 1.20 Excit 0.271 4.44
+#> 3 IGSF3 Inhib 0.899 Excit 0.241 3.72
+#> 4 GALNT14 Inhib 1.86 Excit 0.503 3.69
+#> 5 LYPD6 Inhib 1.09 Astro 0.435 2.51
+#> 6 SLC35D1 Inhib 0.781 OPC 0.375 2.08
+#> 7 FLVCR1 Inhib 0.576 Excit 0.336 1.72
+#> 8 RAVER2 Inhib 1.09 OPC 0.651 1.68
+#> 9 ARHGEF11 Inhib 1.81 Excit 1.10 1.64
+#> 10 VAV3 Inhib 1.68 Astro 1.03 1.63
+#> # ℹ 1,711 more rows
+#> # ℹ 4 more variables: MeanRatio.rank <int>, MeanRatio.anno <chr>,
+#> # gene_ensembl <chr>, gene_name <chr>
+
+## genes with the highest MeanRatio are the best marker genes for each cell type
+mean_ratio_marker_stats |>
+ filter(MeanRatio.rank == 1)
+#> # A tibble: 7 × 10
+#> gene cellType.target mean.target cellType.2nd mean.2nd MeanRatio
+#> <chr> <fct> <dbl> <fct> <dbl> <dbl>
+#> 1 LYPD6B Inhib 1.20 Excit 0.0967 12.4
+#> 2 AC012494.1 Oligo 2.37 OPC 0.147 16.1
+#> 3 MIR3681HG OPC 1.58 Excit 0.205 7.69
+#> 4 AC011995.2 Excit 1.01 Inhib 0.135 7.51
+#> 5 PRDM16 Astro 1.97 EndoMural 0.142 13.9
+#> 6 SLC2A1 EndoMural 1.49 Astro 0.146 10.2
+#> 7 LINC01141 Micro 1.57 Excit 0.0640 24.5
+#> # ℹ 4 more variables: MeanRatio.rank <int>, MeanRatio.anno <chr>,
+#> # gene_ensembl <chr>, gene_name <chr>
+
+
+4. Find 1vALL marker genes
+
+To further explore cell type marker genes it can be helpful to also
+calculate the 1vALL stats for the dataset. To help with this we have
+included the function DeconvoBuddies::findMarkers_1vALL,
+which is a wrapper for scran::findMarkers() that iterates
+through cell types and creates an table output in a compatible with the
+output get_mean_ratio().
+Similarity
+Note this function can take a bit of time to run.
+
+
+marker_stats_1vAll <- findMarkers_1vAll(
+ sce = sce, # sce is the SingleCellExperiment with our data
+ assay_name = "counts",
+ cellType_col = "cellType_broad_hc", # column in colData with cell type info
+ mod = "~BrNum" # Control for donor stored in "BrNum" with mod
+)
+#> 2024-08-12 15:11:00.718289 - Find markers for: Inhib
+#> 2024-08-12 15:12:35.089049 - Find markers for: Oligo
+#> 2024-08-12 15:14:09.079623 - Find markers for: OPC
+#> 2024-08-12 15:15:43.553514 - Find markers for: Excit
+#> 2024-08-12 15:17:17.52289 - Find markers for: Astro
+#> 2024-08-12 15:18:52.162451 - Find markers for: EndoMural
+#> 2024-08-12 15:20:26.036891 - Find markers for: Micro
+#> Building Table - 2024-08-12 15:21:59.434288
+#> ** Done! **
+The function findMarkers_1vALL() returns a
+tibble with the following columns:
+
+-
+
gene is the name of the gene (from
+rownames(sce)).
+-
+
logFC the log fold change from the DE test
+-
+
log.p.value the log of the p-value of the DE test
+-
+
log.FDR the log of the False Discovery Rate adjusted
+p.value
+-
+
std.logFC the standard logFC
+-
+
cellType.target the cell type we’re finding marker
+genes for
+-
+
std.logFC.rank the rank of std.logFC for
+each cell type
+-
+
std.logFC.anno is an annotation of the
+std.logFC value helpful for plotting.
+
+
+## Explore the tibble output
+marker_stats_1vAll
+#> # A tibble: 35,000 × 8
+#> # Groups: cellType.target [7]
+#> gene logFC log.p.value log.FDR std.logFC cellType.target std.logFC.rank
+#> <chr> <dbl> <dbl> <dbl> <dbl> <fct> <int>
+#> 1 SILC1 2.77 -6747. -6739. 1.42 Inhib 1
+#> 2 LYPD6B 2.48 -6338. -6330. 1.37 Inhib 2
+#> 3 LYPD6 2.18 -6147. -6140. 1.35 Inhib 3
+#> 4 ALK 4.56 -5707. -5700. 1.30 Inhib 4
+#> 5 WLS 3.26 -5111. -5104. 1.22 Inhib 5
+#> 6 PLD5 5.85 -4711. -4704. 1.17 Inhib 6
+#> 7 KIRREL1 1.04 -4539. -4532. 1.14 Inhib 7
+#> 8 TRIM67 0.622 -4326. -4319. 1.11 Inhib 8
+#> 9 GREM2 2.11 -4048. -4042. 1.07 Inhib 9
+#> 10 IGSF3 1.27 -3783. -3777. 1.04 Inhib 10
+#> # ℹ 34,990 more rows
+#> # ℹ 1 more variable: std.logFC.anno <chr>
+
+## genes with the highest MeanRatio are the best marker genes for each cell type
+marker_stats_1vAll |>
+ filter(std.logFC.rank == 1)
+#> # A tibble: 7 × 8
+#> # Groups: cellType.target [7]
+#> gene logFC log.p.value log.FDR std.logFC cellType.target std.logFC.rank
+#> <chr> <dbl> <dbl> <dbl> <dbl> <fct> <int>
+#> 1 SILC1 2.77 -6747. -6739. 1.42 Inhib 1
+#> 2 RNF220 10.7 -8373. -8365. 1.52 Oligo 1
+#> 3 BX284613 7.42 -12763. -12754. 4.20 OPC 1
+#> 4 KIAA1211L 7.89 -8371. -8362. 1.23 Excit 1
+#> 5 PRDM16 1.90 -8853. -8844. 2.38 Astro 1
+#> 6 EPAS1 6.50 -9480. -9471. 3.31 EndoMural 1
+#> 7 CSF3R 0.808 -9670. -9661. 3.86 Micro 1
+#> # ℹ 1 more variable: std.logFC.anno <chr>
+
+
+
Reproducibility
@@ -242,7 +490,7 @@ Reproducibility
This package was developed using biocthis.
Code for creating the vignette
-
+
## Create the vignette
library("rmarkdown")
system.time(render("Marker_Finding.Rmd", "BiocStyle::html_document"))
@@ -251,9 +499,9 @@ Reproducibilitylibrary("knitr")
knit("Marker_Finding.Rmd", tangle = TRUE)
Date the vignette was generated.
-#> [1] "2024-08-07 17:35:41 UTC"
+#> [1] "2024-08-12 15:21:59 UTC"
Wallclock time spent generating the vignette.
-#> Time difference of 27.362 secs
+#> Time difference of 11.842 mins
R session information.
#> ─ Session info ───────────────────────────────────────────────────────────────────────────────────────────────────────
#> setting value
@@ -265,8 +513,8 @@ Reproducibility#> collate en_US.UTF-8
#> ctype en_US.UTF-8
#> tz UTC
-#> date 2024-08-07
-#> pandoc 3.2 @ /usr/bin/ (via rmarkdown)
+#> date 2024-08-12
+#> pandoc 3.3 @ /usr/bin/ (via rmarkdown)
#>
#> ─ Packages ───────────────────────────────────────────────────────────────────────────────────────────────────────────
#> package * version date (UTC) lib source
@@ -310,7 +558,7 @@ Reproducibility#> data.table 1.15.4 2024-03-30 [1] RSPM (R 4.4.0)
#> DBI 1.2.3 2024-06-02 [1] RSPM (R 4.4.0)
#> dbplyr 2.5.0 2024-03-19 [1] RSPM (R 4.4.0)
-#> DeconvoBuddies * 0.99.0 2024-08-07 [1] Bioconductor
+#> DeconvoBuddies * 0.99.0 2024-08-12 [1] Bioconductor
#> DelayedArray 0.31.11 2024-08-04 [1] Bioconductor 3.20 (R 4.4.1)
#> desc 1.4.3 2023-12-10 [2] RSPM (R 4.4.0)
#> digest 0.6.36 2024-06-23 [2] RSPM (R 4.4.0)
@@ -334,7 +582,7 @@ Reproducibility#> GenomicAlignments 1.41.0 2024-05-01 [1] Bioconductor 3.20 (R 4.4.0)
#> GenomicRanges * 1.57.1 2024-06-12 [1] Bioconductor 3.20 (R 4.4.0)
#> ggbeeswarm 0.7.2 2023-04-29 [1] RSPM (R 4.4.0)
-#> ggplot2 * 3.5.1 2024-04-23 [1] RSPM (R 4.4.0)
+#> ggplot2 3.5.1 2024-04-23 [1] RSPM (R 4.4.0)
#> ggrepel 0.9.5 2024-01-10 [1] RSPM (R 4.4.0)
#> glue 1.7.0 2024-01-09 [2] RSPM (R 4.4.0)
#> golem 0.4.1 2023-06-05 [1] RSPM (R 4.4.0)
@@ -373,7 +621,7 @@ Reproducibility#> paletteer 1.6.0 2024-01-21 [1] RSPM (R 4.4.0)
#> pillar 1.9.0 2023-03-22 [2] RSPM (R 4.4.0)
#> pkgconfig 2.0.3 2019-09-22 [2] RSPM (R 4.4.0)
-#> pkgdown 2.1.0.9000 2024-08-05 [1] Github (r-lib/pkgdown@1d40a80)
+#> pkgdown 2.1.0.9000 2024-08-12 [1] Github (r-lib/pkgdown@1d40a80)
#> plotly 4.10.4 2024-01-13 [1] RSPM (R 4.4.0)
#> plyr 1.8.9 2023-10-02 [1] RSPM (R 4.4.0)
#> png 0.1-8 2022-11-29 [1] RSPM (R 4.4.0)
@@ -421,8 +669,8 @@ Reproducibility#> SummarizedExperiment * 1.35.1 2024-06-28 [1] Bioconductor 3.20 (R 4.4.1)
#> systemfonts 1.1.0 2024-05-15 [2] RSPM (R 4.4.0)
#> textshaping 0.4.0 2024-05-24 [2] RSPM (R 4.4.0)
-#> tibble * 3.2.1 2023-03-20 [2] RSPM (R 4.4.0)
-#> tidyr * 1.3.1 2024-01-24 [1] RSPM (R 4.4.0)
+#> tibble 3.2.1 2023-03-20 [2] RSPM (R 4.4.0)
+#> tidyr 1.3.1 2024-01-24 [1] RSPM (R 4.4.0)
#> tidyselect 1.2.1 2024-03-11 [1] RSPM (R 4.4.0)
#> timechange 0.3.0 2024-01-18 [1] RSPM (R 4.4.0)
#> UCSC.utils 1.1.0 2024-05-01 [1] Bioconductor 3.20 (R 4.4.0)
diff --git a/pkgdown.yml b/pkgdown.yml
index 69bd4aa..9c8847f 100644
--- a/pkgdown.yml
+++ b/pkgdown.yml
@@ -1,8 +1,8 @@
-pandoc: '3.2'
+pandoc: '3.3'
pkgdown: 2.1.0.9000
pkgdown_sha: 1d40a80e6b3564a6d7da0ce467b0a4570aa5665e
articles:
DeconvoBuddies: DeconvoBuddies.html
Deconvolution_Benchmark_DLPFC: Deconvolution_Benchmark_DLPFC.html
Marker_Finding: Marker_Finding.html
-last_built: 2024-08-07T17:32Z
+last_built: 2024-08-12T15:07Z
diff --git a/reference/dot-get_mean_ratio2.html b/reference/dot-get_mean_ratio2.html
index a2ba3ce..4b8a2dd 100644
--- a/reference/dot-get_mean_ratio2.html
+++ b/reference/dot-get_mean_ratio2.html
@@ -117,7 +117,7 @@ Details
Examples
#' ## load example SingleCellExperiment
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:32:53.195728 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:07:39.587022 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## Get the mean ratio for each gene for each cell type defined in `cellType_broad_hc`
.get_mean_ratio2(sce_DLPFC_example, cellType_col = "cellType_broad_hc")
diff --git a/reference/fetch_deconvo_data.html b/reference/fetch_deconvo_data.html
index 9eb8fbc..806519d 100644
--- a/reference/fetch_deconvo_data.html
+++ b/reference/fetch_deconvo_data.html
@@ -132,9 +132,9 @@ Examples
## A RangedSummarizedExperiment (41.16 MB)
if (!exists("rse-gene")) rse_gene <- fetch_deconvo_data("rse_gene")
-#> 2024-08-07 17:32:55.846502 loading file /github/home/.cache/R/BiocFileCache/4a1de5efa9_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
+#> 2024-08-12 15:07:42.170264 loading file /github/home/.cache/R/BiocFileCache/146b2dbc692f_rse_gene.Rdata%3Frlkey%3Dsw2djr71y954yw4o3xrmjv59b%26dl%3D1
-## explore data
+## explore bulk data
rse_gene
#> class: RangedSummarizedExperiment
#> dim: 21745 110
@@ -147,22 +147,13 @@ Examples
#> 2107UNHS-0291_Br2720_Mid_Cyto ... AN00000906_Br8667_Mid_Cyto
#> AN00000906_Br8667_Mid_Nuc
#> colData names(80): SAMPLE_ID Sample ... diagnosis qc_class
-# class: RangedSummarizedExperiment
-# dim: 21745 110
-# metadata(1): SPEAQeasy_settings
-# assays(2): counts logcounts
-# rownames(21745): ENSG00000227232.5 ENSG00000278267.1 ... ENSG00000210195.2 ENSG00000210196.2
-# rowData names(11): Length gencodeID ... gencodeTx passExprsCut
-# colnames(110): 2107UNHS-0291_Br2720_Mid_Bulk 2107UNHS-0291_Br2720_Mid_Cyto ... AN00000906_Br8667_Mid_Cyto
-# AN00000906_Br8667_Mid_Nuc
-# colData names(78): SAMPLE_ID Sample ... diagnosis qc_class
## load example snRNA-seq data
## A SingleCellExperiment (4.79 MB)
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:32:57.693436 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
-
+#> 2024-08-12 15:07:43.289595 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+## explore example sce data
sce_DLPFC_example
#> class: SingleCellExperiment
#> dim: 557 10000
@@ -176,17 +167,6 @@ Examples
#> reducedDimNames(0):
#> mainExpName: NULL
#> altExpNames(0):
-# class: SingleCellExperiment
-# dim: 557 10000
-# metadata(3): Samples cell_type_colors cell_type_colors_broad
-# assays(2): counts logcounts
-# rownames(557): GABRD PRDM16 ... AFF2 MAMLD1
-# rowData names(7): source type ... gene_type binomial_deviance
-# colnames(10000): 16_CGAAGTTTCGCACGAC-1 11_TTGGGATCAACCGCTG-1 ... 8_CGCATAAGTTAAACCC-1 16_AGCTACATCCCGAGAC-1
-# colData names(32): Sample Barcode ... cellType_layer layer_annotation
-# reducedDimNames(0):
-# mainExpName: NULL
-# altExpNames(0):
## check the logcounts
SingleCellExperiment::logcounts(sce_DLPFC_example)[1:5, 1:5]
@@ -204,6 +184,7 @@ Examples
#> ADGRB2 2.253454 0.0000000
if (FALSE) { # \dontrun{
+## download the full sce experiment object
sce_path_zip <- fetch_deconvo_data("sce")
sce_path <- unzip(sce_path_zip, exdir = tempdir())
sce <- HDF5Array::loadHDF5SummarizedExperiment(
diff --git a/reference/findMarkers_1vAll.html b/reference/findMarkers_1vAll.html
index cb26561..14cec90 100644
--- a/reference/findMarkers_1vAll.html
+++ b/reference/findMarkers_1vAll.html
@@ -76,7 +76,7 @@ Calculate 1 vs. All standard fold change for each gene x cell type, wrapper
assay_name = "counts",
cellType_col = "cellType",
add_symbol = FALSE,
- mod = "~donor",
+ mod = NULL,
verbose = TRUE
)
@@ -102,7 +102,9 @@ Arguments
mod
-String specifying the model used as design in findMarkers. Can be NULL if there are no blocking terms with uninteresting factors as documented at pairwiseTTests.
String specifying the model used as design in findMarkers.
+Can be NULL (default) if there are no blocking terms with uninteresting
+factors as documented at pairwiseTTests.
verbose
@@ -111,28 +113,88 @@ Arguments
Value
- Table of 1 vs. ALL std log fold change + p-values for each gene x cell type
-
+ Tibble of 1 vs. ALL std log fold change + p-values for each gene x cell type
gene is the name of the gene (from rownames(sce)).
logFC the log fold change from the DE test
log.p.value the log of the p-value of the DE test
log.FDR the log of the False Discovery Rate adjusted p.value
std.logFC the standard logFC
cellType.target the cell type we're finding marker genes for
std.logFC.rank the rank of std.logFC for each cell type
std.logFC.anno is an annotation of the std.logFC value
+helpful for plotting.
Examples
- markers_1vAll <- findMarkers_1vAll(sce_ab)
-#> A - '2024-08-07 17:32:58.690142
-#> B - '2024-08-07 17:32:59.008849
-#> Building Table - 2024-08-07 17:32:59.164719
+ ## load example SingleCellExperiment
+if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
+#> 2024-08-12 15:07:45.774661 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+## Explore properties of the sce object
+sce_DLPFC_example
+#> class: SingleCellExperiment
+#> dim: 557 10000
+#> metadata(3): Samples cell_type_colors cell_type_colors_broad
+#> assays(1): logcounts
+#> rownames(557): GABRD PRDM16 ... AFF2 MAMLD1
+#> rowData names(7): source type ... gene_type binomial_deviance
+#> colnames(10000): 8_AGTGACTGTAGTTACC-1 17_GCAGCCAGTGAGTCAG-1 ...
+#> 12_GGACGTCTCTGACAGT-1 1_GGTTAACTCTCTCTAA-1
+#> colData names(32): Sample Barcode ... cellType_layer layer_annotation
+#> reducedDimNames(0):
+#> mainExpName: NULL
+#> altExpNames(0):
+
+## this data contains logcounts of gene expression
+SummarizedExperiment::assays(sce_DLPFC_example)$logcounts[1:5, 1:5]
+#> 8_AGTGACTGTAGTTACC-1 17_GCAGCCAGTGAGTCAG-1 3_CTGGACGAGCTTCATG-1
+#> GABRD 0 0.9249246 0.000000
+#> PRDM16 0 0.0000000 0.000000
+#> MICOS10 0 0.0000000 0.000000
+#> LINC01141 0 0.0000000 0.000000
+#> ADGRB2 0 0.9249246 2.253612
+#> 13_CCCTCAAAGTCTAGCT-1 11_TGTAAGCCATTCTGTT-1
+#> GABRD 0.000000 0.0000000
+#> PRDM16 0.000000 0.0000000
+#> MICOS10 0.000000 0.6528615
+#> LINC01141 0.000000 0.0000000
+#> ADGRB2 2.253454 0.0000000
+
+## nuclei are classified in to cell types
+table(sce_DLPFC_example$cellType_broad_hc)
+#>
+#> Astro EndoMural Micro Oligo OPC Excit Inhib
+#> 692 417 316 1970 350 4335 1920
+
+## Get the 1vALL stats for each gene for each cell type defined in `cellType_broad_hc`
+marker_stats_1vAll <- findMarkers_1vAll(
+ sce = sce_DLPFC_example,
+ assay_name = "logcounts",
+ cellType_col = "cellType_broad_hc",
+ mod = "~BrNum"
+)
+#> 2024-08-12 15:07:46.02474 - Find markers for: Oligo
+#> 2024-08-12 15:07:46.488802 - Find markers for: Excit
+#> 2024-08-12 15:07:46.874934 - Find markers for: Inhib
+#> 2024-08-12 15:07:47.995957 - Find markers for: Micro
+#> 2024-08-12 15:07:48.331533 - Find markers for: OPC
+#> 2024-08-12 15:07:48.667361 - Find markers for: EndoMural
+#> 2024-08-12 15:07:49.036291 - Find markers for: Astro
+#> Building Table - 2024-08-12 15:07:49.415034
#> ** Done! **
-head(markers_1vAll)
+
+## explore output, top markers have high logFC
+head(marker_stats_1vAll)
#> # A tibble: 6 × 8
-#> # Groups: cellType.target [2]
-#> gene logFC log.p.value log.FDR std.logFC cellType.target rank_marker
-#> <chr> <dbl> <dbl> <dbl> <dbl> <chr> <int>
-#> 1 G-D1_A 0.5 -3.65e+ 1 -3.58e+ 1 1.97 A 1
-#> 2 G-D2_A 0.5 -3.65e+ 1 -3.58e+ 1 1.97 A 2
-#> 3 G-D1_B -0.5 -1.42e-16 -1.42e-16 -1.97 A 3
-#> 4 G-D2_B -0.5 -1.42e-16 -1.42e-16 -1.97 A 4
-#> 5 G-D2_B 0.5 -3.65e+ 1 -3.58e+ 1 1.97 B 1
-#> 6 G-D1_B 0.5 -3.65e+ 1 -3.58e+ 1 1.97 B 2
-#> # ℹ 1 more variable: anno_logFC <chr>
+#> # Groups: cellType.target [1]
+#> gene logFC log.p.value log.FDR std.logFC cellType.target std.logFC.rank
+#> <chr> <dbl> <dbl> <dbl> <dbl> <fct> <int>
+#> 1 ST18 4.46 -6697. -6691. 4.32 Oligo 1
+#> 2 PLP1 4.25 -5233. -5227. 3.50 Oligo 2
+#> 3 MOBP 3.18 -4821. -4816. 3.28 Oligo 3
+#> 4 ENPP2 2.72 -4521. -4516. 3.12 Oligo 4
+#> 5 TF 3.00 -4466. -4462. 3.09 Oligo 5
+#> 6 RNF220 3.80 -4390. -4385. 3.05 Oligo 6
+#> # ℹ 1 more variable: std.logFC.anno <chr>
diff --git a/reference/get_mean_ratio.html b/reference/get_mean_ratio.html
index e96dc51..c99630d 100644
--- a/reference/get_mean_ratio.html
+++ b/reference/get_mean_ratio.html
@@ -121,8 +121,10 @@ Value
mean.2nd is the mean expression of gene for cellType.2nd.
MeanRatio is the ratio of mean.target/mean.2nd.
MeanRatio.rank is the rank of MeanRatio for the cell type.
MeanRatio.anno is an annotation of the MeanRatio calculation helpful for plotting.
gene_ensembl & gene_name optional cols spcified by the user to add gene infomation
MeanRatio.anno is an annotation of the MeanRatio calculation helpful
+for plotting.
gene_ensembl & gene_name optional cols from rowData(sce)specified by
+the user to add gene information
Details
@@ -134,7 +136,7 @@ Details
Examples
## load example SingleCellExperiment
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:00.419412 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:07:50.863967 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## Explore properties of the sce object
sce_DLPFC_example
#> class: SingleCellExperiment
@@ -221,7 +223,10 @@ Examples
#> MAMLD1 protein_coding 75492.1
## specify rowData col names for gene_name and gene_ensembl
-get_mean_ratio(sce_DLPFC_example, cellType_col = "cellType_broad_hc", gene_name = "gene_name", gene_ensembl = "gene_id")
+ get_mean_ratio(sce_DLPFC_example,
+ cellType_col = "cellType_broad_hc",
+ gene_name = "gene_name",
+ gene_ensembl = "gene_id")
#> # A tibble: 762 × 10
#> gene cellType.target mean.target cellType.2nd mean.2nd MeanRatio
#> <chr> <fct> <dbl> <fct> <dbl> <dbl>
diff --git a/reference/plot_gene_express.html b/reference/plot_gene_express.html
index 413056c..1b3d700 100644
--- a/reference/plot_gene_express.html
+++ b/reference/plot_gene_express.html
@@ -144,7 +144,7 @@ Examples
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:06.529501 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:07:56.587053 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## plot expression of two genes
plot_gene_express(sce = sce_DLPFC_example, category = "cellType_broad_hc", genes = c("GAD2", "CD22"))
#> No summary function supplied, defaulting to `mean_se()`
diff --git a/reference/plot_marker_express.html b/reference/plot_marker_express.html
index 9dc7169..dc08ee0 100644
--- a/reference/plot_marker_express.html
+++ b/reference/plot_marker_express.html
@@ -155,7 +155,7 @@ See also
Examples
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:09.904147 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:07:59.949517 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## plot the top markers for Astrocytes in
plot_marker_express(
sce = sce_DLPFC_example,
diff --git a/reference/plot_marker_express_ALL.html b/reference/plot_marker_express_ALL.html
index 6749dc8..1e360ad 100644
--- a/reference/plot_marker_express_ALL.html
+++ b/reference/plot_marker_express_ALL.html
@@ -153,7 +153,7 @@ See also
Examples
#' ## Fetch sce example data
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:12.916615 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:08:02.934026 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
# Plot marker gene expression to PDF, one page per cell type in stats
pdf_file <- tempfile("test_marker_expression_ALL", fileext = ".pdf")
diff --git a/reference/plot_marker_express_List.html b/reference/plot_marker_express_List.html
index afdac08..1ce1204 100644
--- a/reference/plot_marker_express_List.html
+++ b/reference/plot_marker_express_List.html
@@ -134,7 +134,7 @@ See also
Examples
## Fetch sce example data
if (!exists("sce_DLPFC_example")) sce_DLPFC_example <- fetch_deconvo_data("sce_DLPFC_example")
-#> 2024-08-07 17:33:29.315545 loading file /github/home/.cache/R/BiocFileCache/4a1ec11882_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
+#> 2024-08-12 15:08:19.448584 loading file /github/home/.cache/R/BiocFileCache/146b342eafd5_sce_DLPFC_example.Rdata%3Frlkey%3Dv3z4u8ru0d2y12zgdl1az07q9%26st%3D1dcfqc1i%26dl%3D1
## Create list-of-lists of genes to plot, names of sub-list become title of page
my_gene_list <- list(Inhib = c("GAD2", "SAMD5"), Astro = c("RGS20", "PRDM16"))