[Deprecated: See Readme] SmartSeq2 Pipeline (v0.1)
The pipeline documentation is now kept up to date and released alongside the pipeline in a readme. Please navigate to the latest Smartseq2 Release in Github and then to /pipelines/smartseq2_single_sample
Welcome to HCA Skylab scRNA-Seq pipeline page. Using this page will navigate you through our first draft of full-transcriptome-length scRNA-Seq pipeline.
This pipeline is designed to process scRNA-Seq data in Google cloud computing environment.
This pipeline is written in Cromwell/WDL.
There are several Dockers have been built for this pipeline.
This dataset was published in this paper "Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma" . A subset of the full dataset were downloaded from GSE57872.
Obtain a reference genome from Ensembl, UCSC or NCBI. We use the human genome reference hg19 downloaded from UCSC in our draft workflow. Further more, we only use a single chromosome (chr21) for our demo workflow.
Genome annotation is obtained from Gencode. We downloaded v19 gtf file. We also download refFlat file from UCSC.
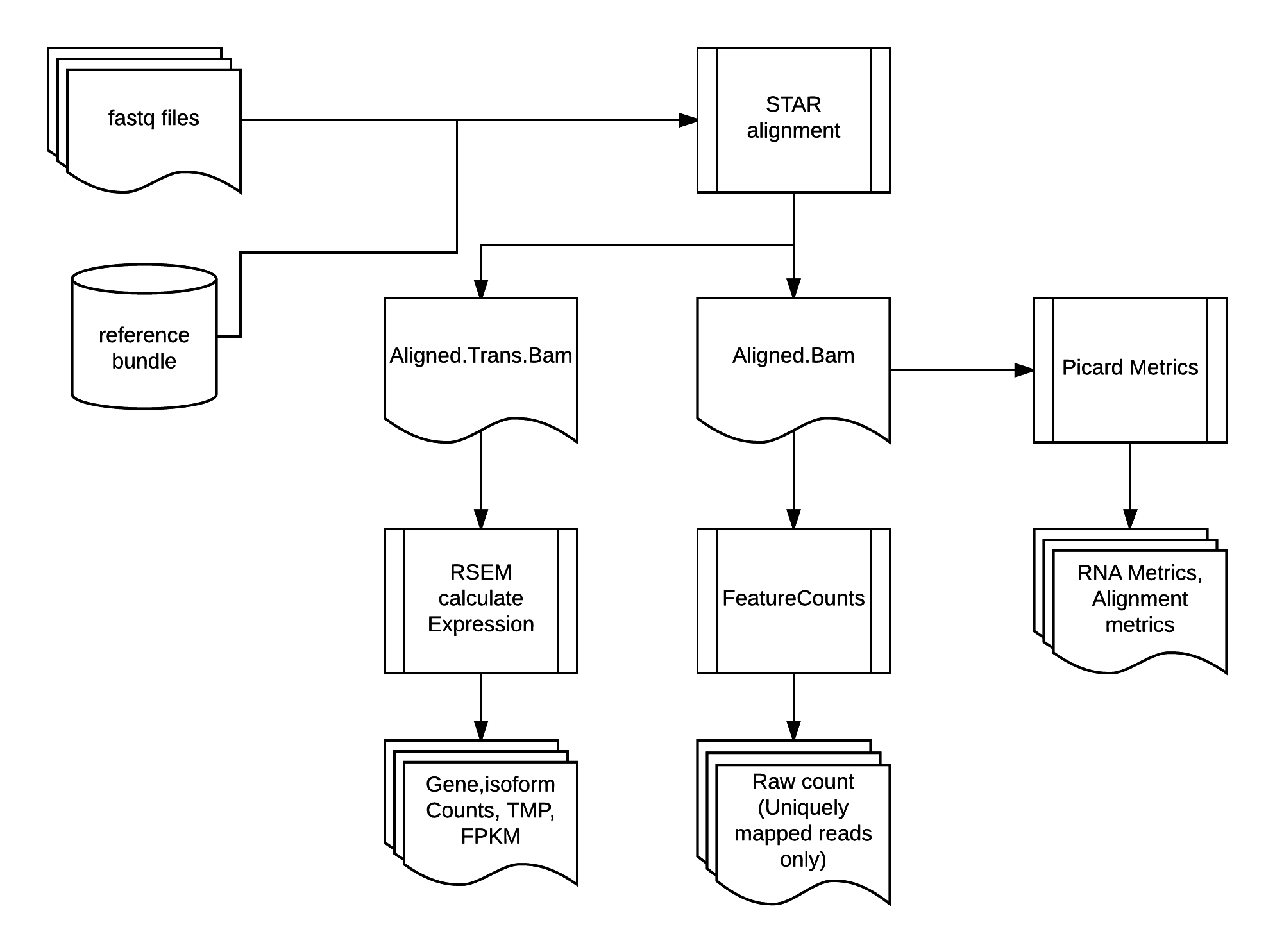
Perform the alignment with STAR to align fastq reads to genome and transcriptome. Reference indexes are built for STAR and RSEM pre-alignment. For downstream quantification purpose, besides genome aligned bam file, we also output transcriptome aligned bam file from STAR alignment run.
RSEM perform expected counts quantification on gene/isoform features from transcriptome aligned bam file. FeatureCounts perform raw counts quantification on gene/exon/trancript features from genome aligned bam file by only using uniquely mapped reads.
Picard collects two post-alignment QC metrics. AlignmentSummaryMetrics is collected by
CollectAlignmentSummaryMetrics module. RnaSeqMetrics is collected by CollectRnaSeqMetrics module.
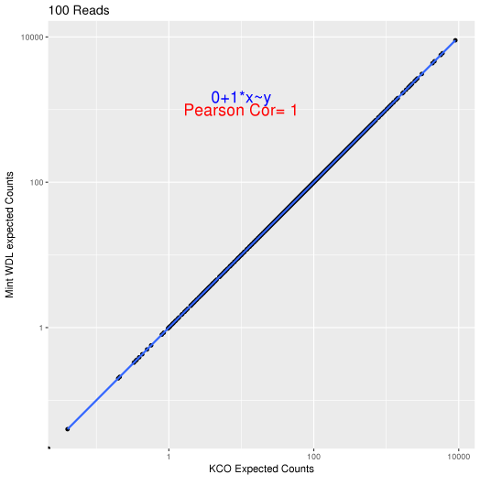
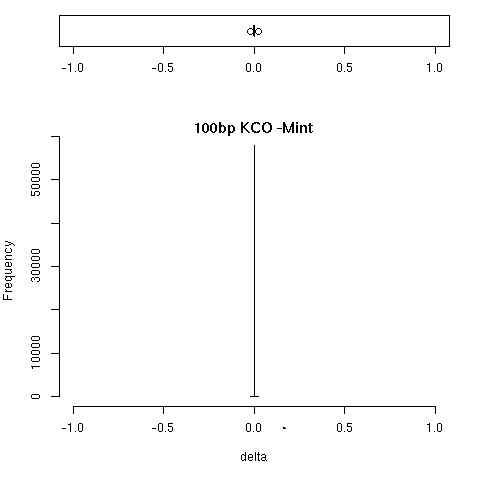
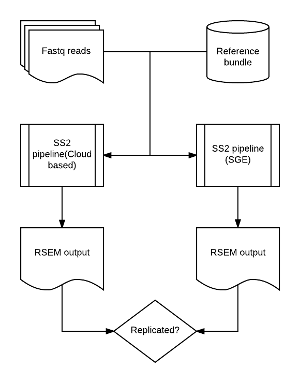
To replicate pipeline results between running environments. Our pipeline is up running in cloud-based computing environment and we try to replicate output of the same pipeline but execute the pipeline in local SGE computing environment. Given the same input fastq files and reference builds, pipeline are executed in both google cloud and local SGE. Then we compare the feature's quantification results, such as RSEM expected counts results.
We calculated the correlation between RSEM's expected counts from individual two pipeline executions. We also calculated the difference between these two RSEM's expected counts.