[Deprecated: See Readme] SmartSeq2 Pipeline (v0.2.0)
The pipeline documentation is now kept up to date and released alongside the pipeline in a readme. Please navigate to the latest Smartseq2 Release in Github and then to /pipelines/smartseq2_single_sample
- scRNASeq Pipeline Overview
- SmartSeq2 scRNASeq Quality Control
- SmartSeq2 scRNASeq Quantification
- Enviroment
- QC Reports
The full length SMART-seq 2 scRNASeq pipeline is designed to process data submitted to the Human Cell Atlas. The pipeline is written in WDL, is freely available on Github, and can be run by any compliant WDL runner (e.g. crowmell). The pipeline is designed to process stranded or un-stranded paired-ended scRNA-seq data.
The pipeline include two modules, a Quality Control module and a Transcriptome Quantification module. The Quality Control Module generates a set of post alignment quality control metrics. The Transcriptome Quantification Module aligns reads to the genome and estimates transcript expression levels. The inputs of pipeline are paired-end fastq files (fastq.gz) and a pre-built reference bundle. This file shows an example input. The pipeline WDL is found here. The below figure summarizes the execution graph of the pipeline.
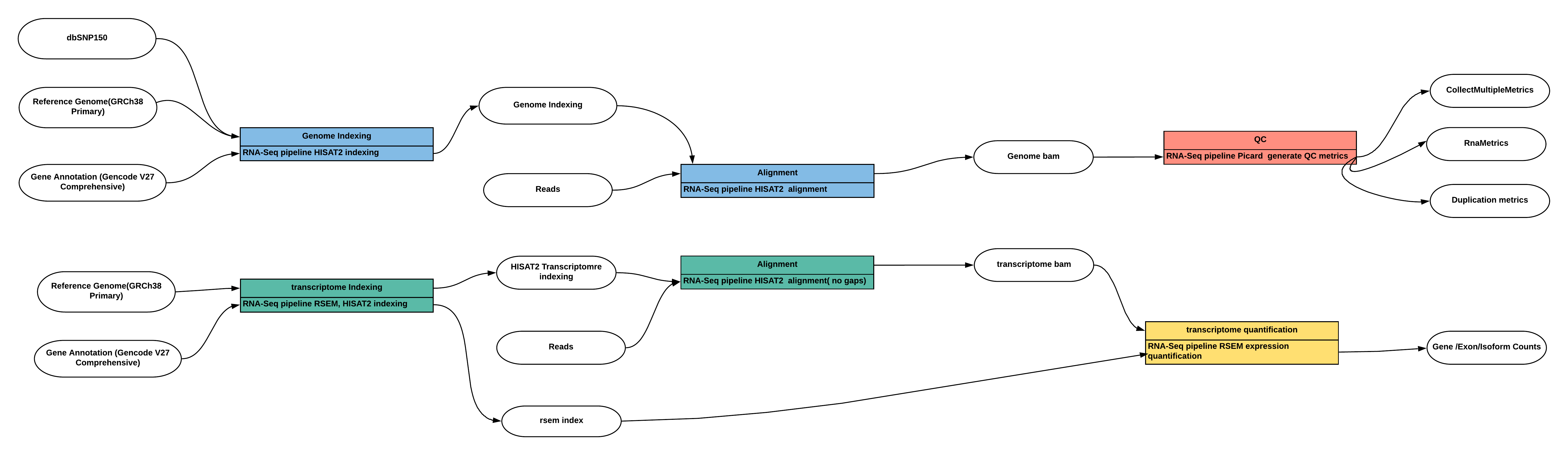 View the full size figure of this pipeline schematics
View the full size figure of this pipeline schematics
{kind=link}
In this section, we will describe each step in the Quality Control (QC) module. The QC workflow WDL file is available at Github.
We obtain the Genome sequence primary assembly(GRCh38) and the Comprehensive gene annotation from Gencode. The version of Gencode annotation used in the workflow is v27.
The HCA Smart-seq2 pipeline currently supports mouse reference and gene annotation from Gencode. The specific mouse reference is the Genome Reference Consortium Mouse Build 38 Organism name: Mus musculus (C57BL/6J).
HISAT2 can incorporate exons, splice sites and single nucleotide polymorphism (SNP) locations into the index file for alignment. HISAT2 also provides a set of python scripts to build an index using Ensembl or RefSeq annotations. We modify this script to construct an index with the Gencode annotation. The workflow to build the reference index is here.
make_grch38_snp_tran_gencode.sh ${gtf_version} ${dbsnp_version}
mkdir ${ref_name}
cp *.ht2 ${ref_name}
tar -zcvf "${ref_name}.tar.gz" "${ref_name}"To learn more about how the HISAT2 indexing strategy is distinct from other next generation aligners refer to the HISAT publication.
RSEM provides a program to extract and pre-process the reference sequences, then build its index. The workflow to build the RSEM index is available at Github.
mkdir rsem
rsem-prepare-reference --gtf ${gtf_file} --bowtie ${ref_fasta} rsem/rsem_trans_index
tar -cvf ${ref_name}.tar rsem/To learn more about how to build the RSEM index, refer to the RSEM website. Note A transcriptome reference Fasta file is included in RSEM index bundle. This file will be used to create the HISAT2 transcriptome index.
To build a trancriptome index for HISAT2, we can run the following command:
hisat2-build -p 8 rsem/rsem_trans_index.idx.fa output_nameThe workflow to build the transcriptome index for HISAT2 is available here.
HISAT2 uses a graph-based alignment and has succeeded HISAT, Tophat2 and Bowtie2. The output of HISAT2 alignment workflow is a bam file.
HISAT2 can perform alignments to the genome or transcriptome. In this pipeline, we use the following command line to execute HISAT2 alignment.
hisat2 -t \
-x ${ref_name}/${ref_name} \
-1 ${fq1} \
-2 ${fq2} \
--rg-id=${sample_name} --rg SM:${sample_name} --rg LB:${sample_name} \
--rg PL:ILLUMINA --rg PU:${sample_name} \
--new-summary --summary-file ${output_name}.log \
--met-file ${output_name}.hisat2.met.txt --met 5 \
--seed 12345 \
-k 10 \
--secondary \
-p 4 -S ${output_name}.sam Details of options specified below:
-
-p 4tells HISAT2 to use eight CPUs for bowtie alignments. -
--rg-id ${sample_name}specifies a read group ID that is a unique identifier. -
--rg SM:${sample_name}specifies a read group sample name. This together with rg-id will allow you to determine which reads came from which sample in the merged bam later on. -
--rg LB:${sample_name}specifies a read group library name. This together with rg-id will allow you to determine which reads came from which library in the merged bam later on. -
--rg PL:ILLUMINAspecifies a read group sequencing platform. -
--rg PU:$PLATFORM_UNITspecifies a read group sequencing platform unit. Typically this consists of FLOWCELL-BARCODE.LANE -
-x ${ref_name}/${ref_name}The HISAT2 index filename prefix (minus the trailing .X.ht2) built earlier including splice sites and exons. -
-1 ${fq1}The read 1 FASTQ file, optionally gzip(.gz) or bzip2(.bz2) compressed. -
-2 ${fr2}The read 2 FASTQ file, optionally gzip(.gz) or bzip2(.bz2) compressed. -
-S ${output_name}.samThe output SAM format text file of alignments. -
--seed 12345To fix a pseudo-random seed number in order to outputdeterministicalignment. -
-k 10To output up to 10 secondary alignemnts. Default value is10 -
--secondaryAllow to output secondary alignments. DefaultOn -
--new-summaryTo output HISAT2 alignment summary into a log file with machine friendly format -
--met-file ${output_name}.hisat2.met.txt --met 5Write metric file.
Note In our pipeline, we treat each input as single library. HISAT2 only outputs alignments to a SAM file. We need to convert SAM to BAM by using Samtools, and we sort by alignment position to reduce stored file size.
samtools sort -@ 4 -O bam -o "${output_name}.bam" "${output_name}.sam" HISAT2 generates a summary of the alignments printed to the terminal or a log file. The example is shown as following.
HISAT2 summary stats:
Total pairs: 708464
Aligned concordantly or discordantly 0 time: 143976 (20.32%)
Aligned concordantly 1 time: 476798 (67.30%)
Aligned concordantly >1 times: 85899 (12.12%)
Aligned discordantly 1 time: 1791 (0.25%)
Total unpaired reads: 287952
Aligned 0 time: 182477 (63.37%)
Aligned 1 time: 67717 (23.52%)
Aligned >1 times: 37758 (13.11%)
Overall alignment rate: 87.12%Note Total unpaired reads: 287952 is doubled of Aligned concordantly or discordantly 0 time: 143976. In HISAT2, the concordantly of paired-end alignment means two reads from a pair must be mapped and within proper distance. All the other cases are counted as discordantly
[Background] Picard is a set of command line tools (in Java) for manipulating high-throughput sequencing data and also can be used to generate QC reports for RNA-seq.
Here is a list of QC metrics generated by Picard which are collected in our pipeline:
- CollectAlignmentSummaryMetrics
- CollectRnaSeqMetrics
- MarkDuplicates
- CollectInsertSizeMetrics
- CollectGcBiasMetrics
- CollectBaseDistributionByCycle
- QualityScoreDistribution
- MeanQualityByCycle
- CollectSequencingArtifactMetrics
- CollectQualityYieldMetrics
Files needed to run Picard:
- Aligned or un-aligned bam files. Additional files are required for specific metrics:
- A reference fasta file.
- Ribosomal interval file.
- genePred's
refFlatfile.
To convert gene annotations from Gencode GTF to refFlat format, execute the following:
gtfToGenePred -genePredExt -geneNameAsName2 genes.gtf refFlat.tmp.txt
paste <(cut -f 12 refFlat.tmp.txt) <(cut -f 1-10 refFlat.tmp.txt) > refFlat.txt
rm refFlat.tmp.txtgtfToGenePred can be obtained from http://hgdownload.cse.ucsc.edu/admin/exe/
To convert gene annotation from Gencode GTF to Interval_list format:
chrom_sizes='chrm_sizes.txt'
samtools view -H $aligned_bam > $chrom_sizes
genes='gencode.v27.gtf'
rRNA='rRNA.interval_list'
# Sequence names and lengths. (Must be tab-delimited.): only output SQ tag
perl -lane 'print "\@SQ\tSN:$F[0]\tLN:$F[1]\tAS:hg19"' $chrom_sizes | \
grep -v _ \
>> $rRNA
# Intervals for rRNA transcripts.
grep 'gene_type "rRNA"' $genes | \
awk '$3 == "transcript"' | \
cut -f1,4,5,7,9 | \
perl -lane '
/transcript_id "([^"]+)"/ or die "no transcript_id on $.";
print join "\t", (@F[0,1,2,3], $1)
' | \
sort -k1V -k2n -k3n \
>> $rRNAThe pipeline uses RSEM to generate expression estimates from the SAM/BAM files generated by HISAT2 alignment of reads against a transcriptome. The quantification workflow is available here.
Performs alignment with HISAT2 to transcriptome:
hisat2 -t \
-x ${ref_name}/${ref_name} \
-1 ${fq1} \
-2 ${fq2} \
--rg-id=${sample_name} --rg SM:${sample_name} --rg LB:${sample_name} \
--rg PL:ILLUMINA --rg PU:${sample_name} \
--new-summary --summary-file ${output_name}.log \
--met-file ${output_name}.hisat2.met.txt --met 5 \
-k 10 \
--mp 1,1 \
--np 1 \
--score-min L,0,-0.1 \
--secondary \
--no-mixed \
--no-softclip \
--no-discordant \
--rdg 99999999,99999999 \
--rfg 99999999,99999999 \
--no-spliced-alignment \
--seed 12345 \
-p 4 -S ${output_name}.sam The specific parameters for the transcriptome alignment are:
-
-x ${ref_name}/${ref_name}this is transcriptome index files which is built from RSEM extractedtranscript.fa. -
--mp 1,1reduce mis-matching panel to 1 for all regardless how many of mis-matching there are. -
np 1reduceNbases panel to 1 as well. -
--score-min L,0,-0.1initial the mapping score. -
--no-mixedand--no-discordantno partial alignment allow and only output properly aligned paired-end reads. -
--no-softclipno soft clip alignment allowed. -
--rdg 99999999,99999999and--rfg 99999999,99999999Add gap alignment panel to be infinity. -
--no-spliced-alignmentNo splicing alignment allowed.
Then Samtools is applied to convert from SAM to BAM format.
samtools view -bS "${output_name}.sam" > "${output_name}.bam"Note No sorting required if the bam file is a input of RSEM.
The quantification of genes and transcripts is done with the RSEM program. The program rsem-calculate-expression in the RSEM package is used to estimate gene/isoform expression levels. The output can be expected_counts, TPM or FPKM. The command line to run this program is as follows:
rsem-calculate-expression \
--bam \
--paired-end \
-p 4 \
--time --seed 555 \
--calc-pme \
--single-cell-prior \
${trans_aligned_bam} \
rsem/rsem_trans_index \
"${rsem_out}" Details of options specified below:
-
-p 4multiple thread mode, use 4 core. -
--bamand--paired-endInput is aligned paired-end bam file; no alignment will be done by RSEM. -
--timereport running time -
--sed 555fix random seed to produce deterministic results. -
--calc-pmeand--single-cell-priorBeside the MLE estimation, RSEM also produce prior-mean-estimation with single-cell-prior.
Note The -calc-pme and --single-cell-prior has not been thoroughly investigated with single cell data yet so the pme estimation should be used with care.
The main output of rsem-calculate-expression is the rsem.gene.results and rsem.isoform.results which include expected_count,TPM and FPKM
Our pipeline has been designed and tested in the Google Cloud. The applications and software packages required by this pipeline have been pre-built into docker images and are available at Quay Docker Registry. Here is a list of docker images and application required by this pipeline.
Here is a QC report example