SmartSeq2 Pipeline Timing and Cost
It could be very complicated to estimate the cost of cloud computing. For google cloud, the billing is on the reserved resource, such as number of cores, size of harddrive and RAM. You will be charged by a hourly rate over the time you use the reserved resource. The pricing also varies between machine type. For example, the hourly rate of using a high specs machine (more nodes and high RAM) is higher than low specs ones. The pricing page lists details.
The cost of processing RNA-Seq on cloud computing depends one several factors. One is running time. Second how much resource has been reserved during data processing. Third is machine type. Itcould be tricky to estiamte the cost. For example, to run one RSEM job, we can either request min resouce, such as 1-core with 4Gb RAM machine or we can request a 4-core and 15Gb machine. The hourly rate of the first type machine is cheaper than the second one but it takes longer time to finish job.
In this task, we estimated the cost of processing scRNA-Seq data by using google cloud. The testing pipeline included the following modules/steps:
- STAR alignment [request 8-core and 40Gb RAM]
- RSEM to estimate gene counts [request 4-core and 4Gb RAM]
- FeatureCount to calculate gene/exon/transcript counts [request 1-core and 4Gb RAM]
- Picard to collect several sequencing and RNA-Seq specific metrics [request 1-core and 4Gb RAM]
- Python script to parse pipeline output [request 1-core and 2Gb]
Then we tested this pipeline on a published scRNA-Seq dataset, which includes 864 single cells full length RNA-Seq data. And we summaried the total hours(in hour) and total cost(in dollar) in the following table.
| StepName | Program/software | Machine Type | Total Hours | Total Cost | AVG Hours | AVG Cost |
|---|---|---|---|---|---|---|
| Star | Star | n1-highmem-8 | 292.133 | 140.115 | 0.338 | 0.1621 |
| RSEM | RSEM | n1-standard-4 | 151.983 | 29.376 | 0.1759 | 0.034 |
| CollectAlignemntMetrics | Picard | n1-standard-2 | 144.216 | 13.858 | 0.166 | 0.016 |
| CollectRnaMetrics | Picard | n1-standard-2 | 144.555 | 13.890 | 0.167 | 0.0160 |
| CollectInsertSizeMetrics | Picard | n1-standard-2 | 144.116 | 13.849 | 0.1668 | 0.016 |
| CollectDuplicationMetrics | Picard | n1-standard-2 | 144.433 | 13.958 | 0.167 | 0.0161 |
| FeatureCountsUniqueCounts | FeatureCount | n1-standard-2 | 144.266 | 14.179 | 0.167 | 0.0164 |
| FeatureCountsMultiCounts | FeatureCount | n1-standard-2 | 144.483 | 14.200 | 0.1672 | 0.0164 |
| CollectMetricsbySample | python | n1-standard-1 | 144.466 | 7.535 | 0.167 | 0.0087 |
We also examed the impact fo scRNA-Seq data quality on total hours and total cost. The two QC measurements we examed are TOTAL READS and PCT_USABLE_BASES. In general, the first measure would tell us the size of data and second measurement would tell us the overall quality of RNA-Seq experiement.
For example,we examed Star timing and cost with READ_LENGTH,TOTAL READS and PCT_USABLE_BASES
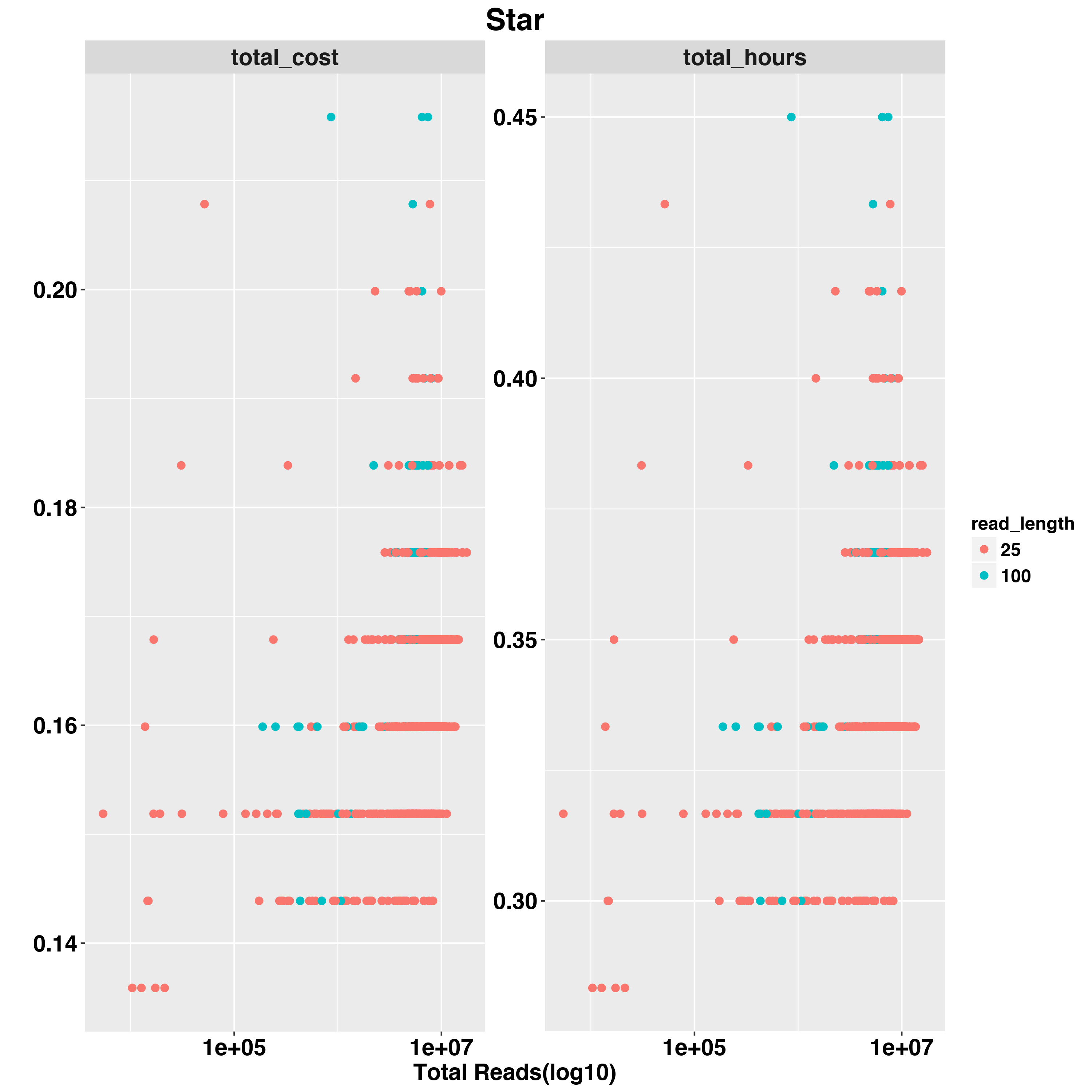
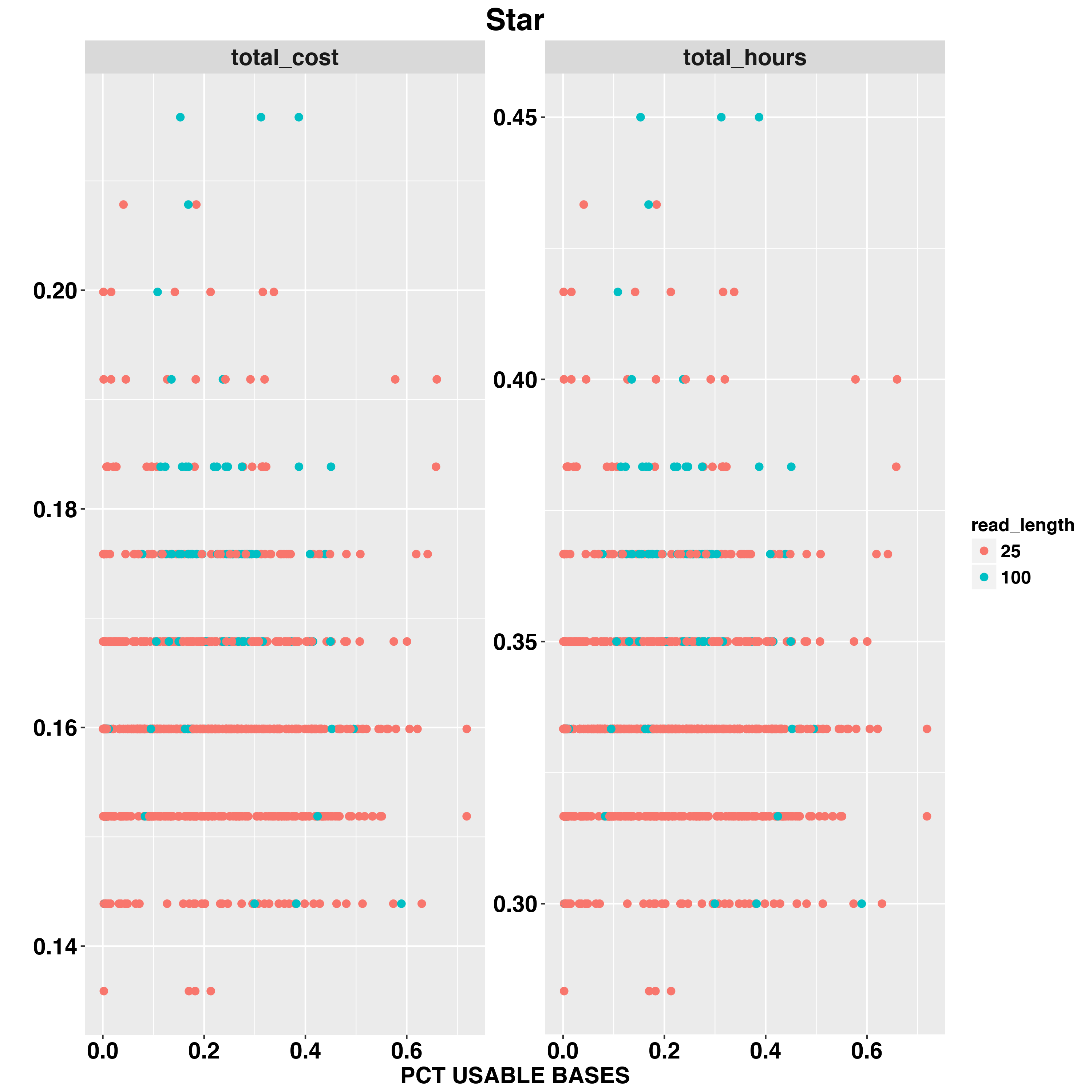
And RSEM


And FeatureCounts


The Star and RSEM will run longer with larger input sequencing data. RSEM and FeatureCounts are both quantification methods but FeaetureCounts is less impacted by input data size. PCT_USABLE_BASES has less impact on timing and cost compared to TOTAL_READS.